
Insights into Human Astrocyte Response to H5N1 Infection by Microarray Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
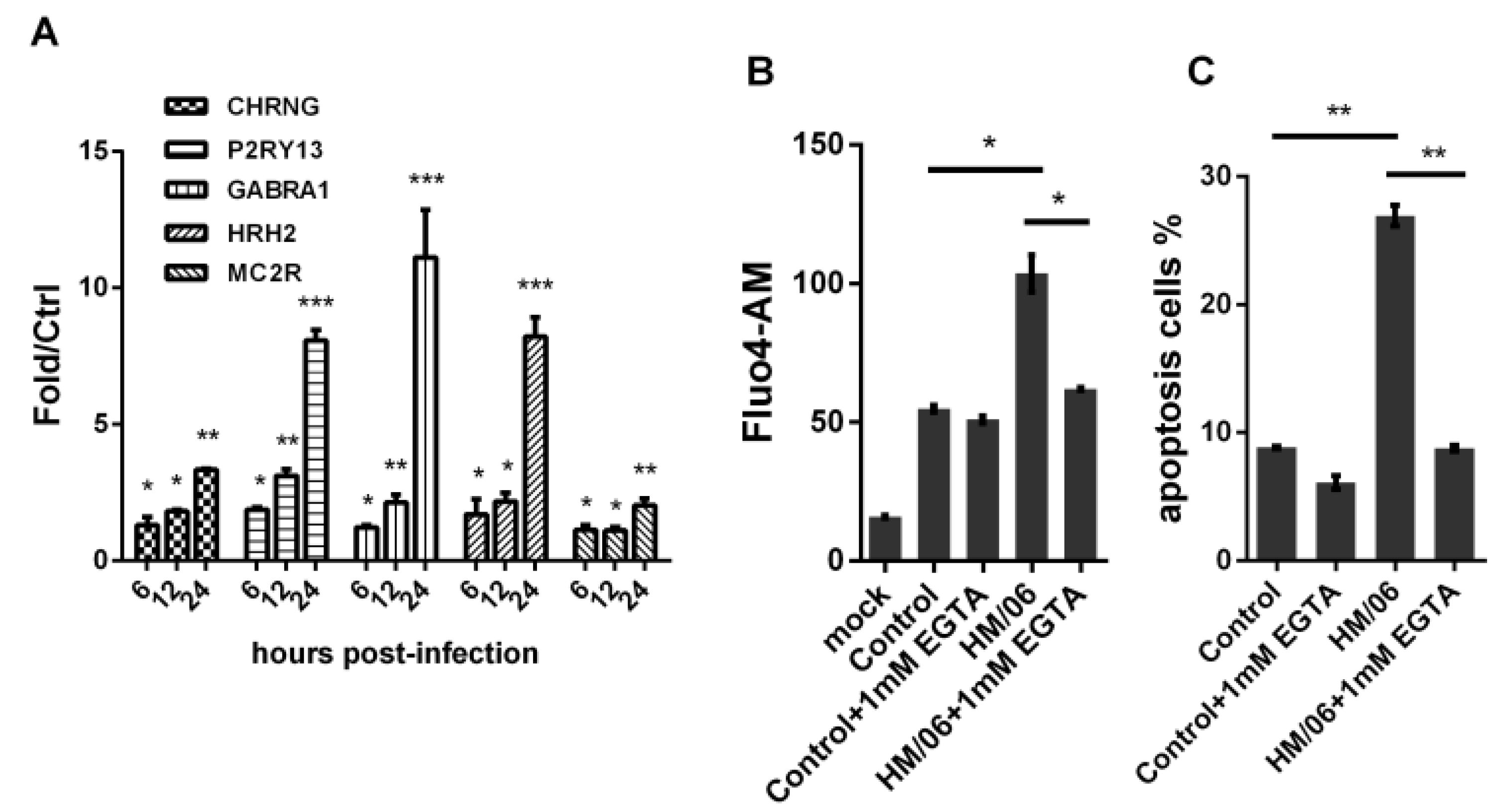
2.2. Apoptosis Detection
2.3. Immunofluorescence Assay
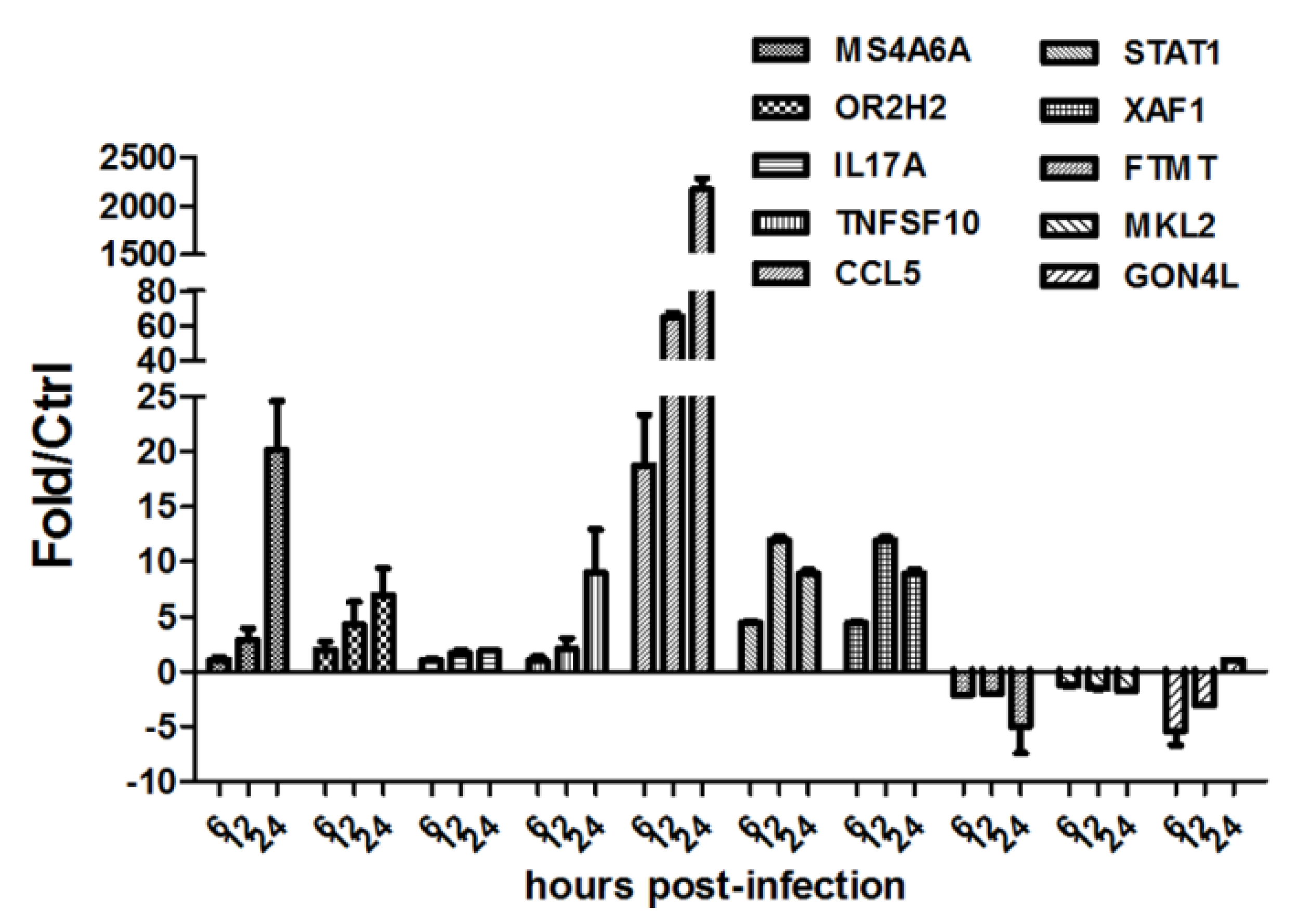
2.4. Agilent Microarray Experiments
2.5. Western Blot
2.6. Real-Time Quantitative RT-PCR (qRT-PCR) Assays
2.7. Detection of Ca2+Accumulation
2.8. Statistical Analysis
3. Results
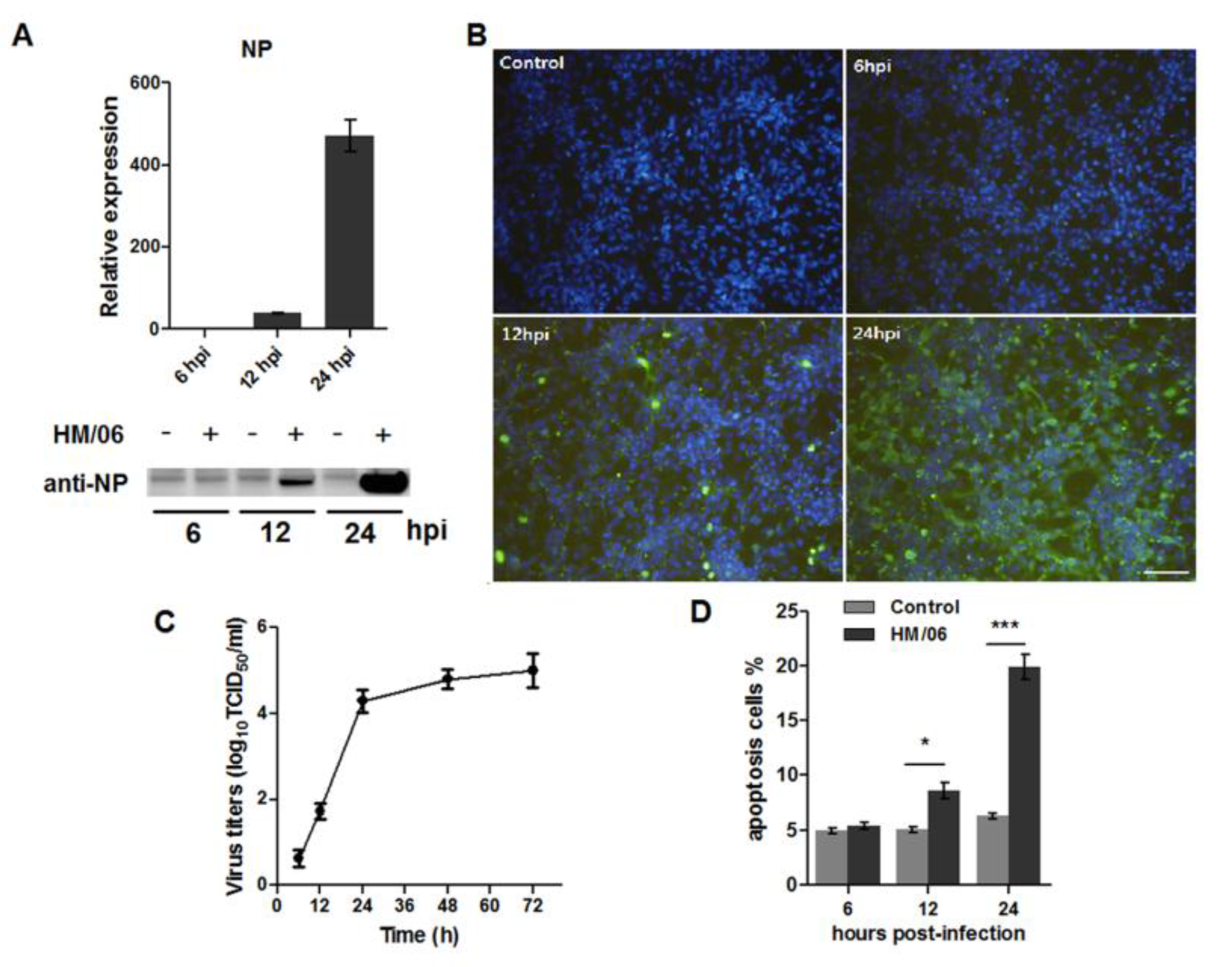
3.1. Infection of U251 Cells by HM/06
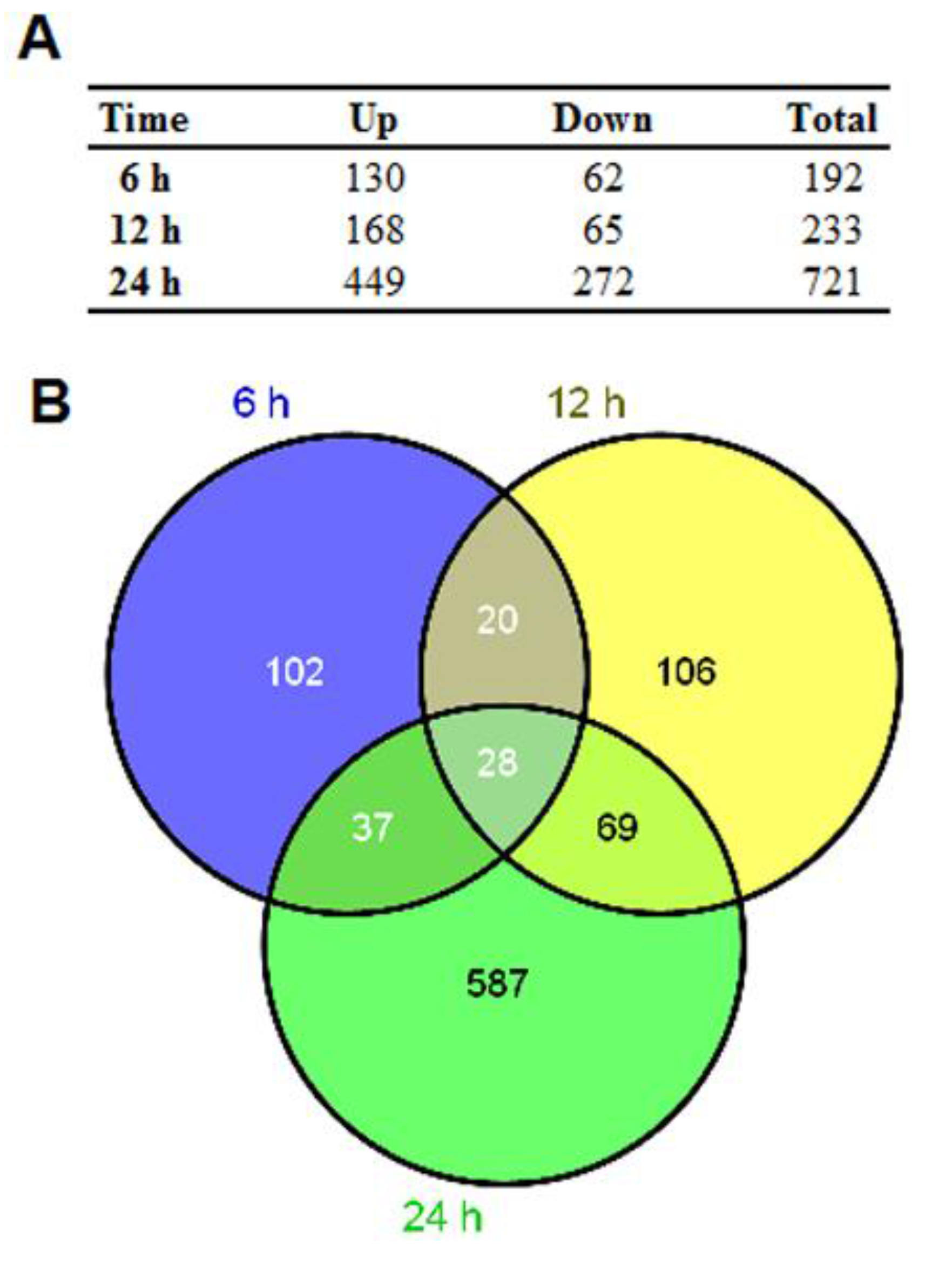
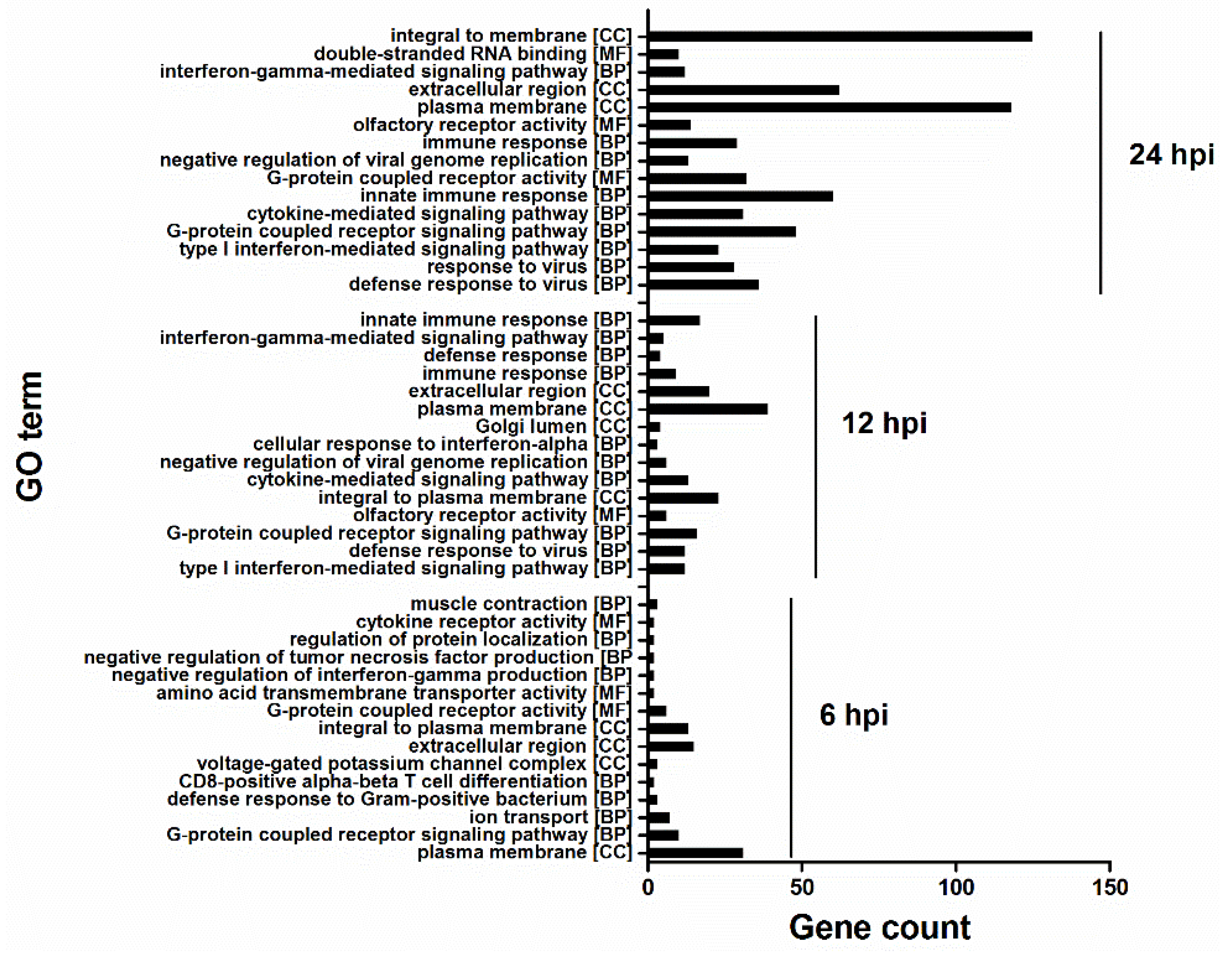
3.2. Gene Expression Alterations in Infected U251 Cells





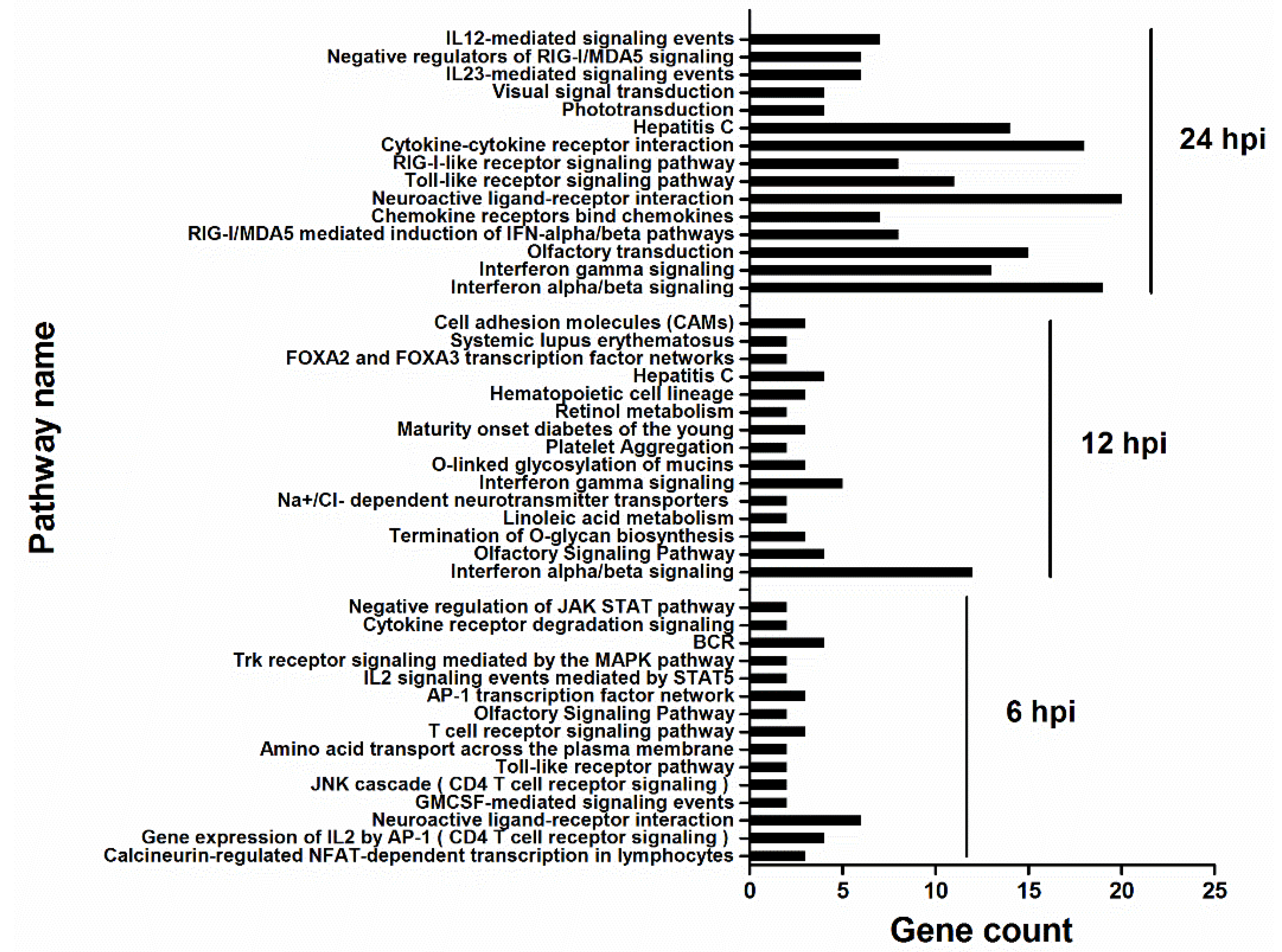
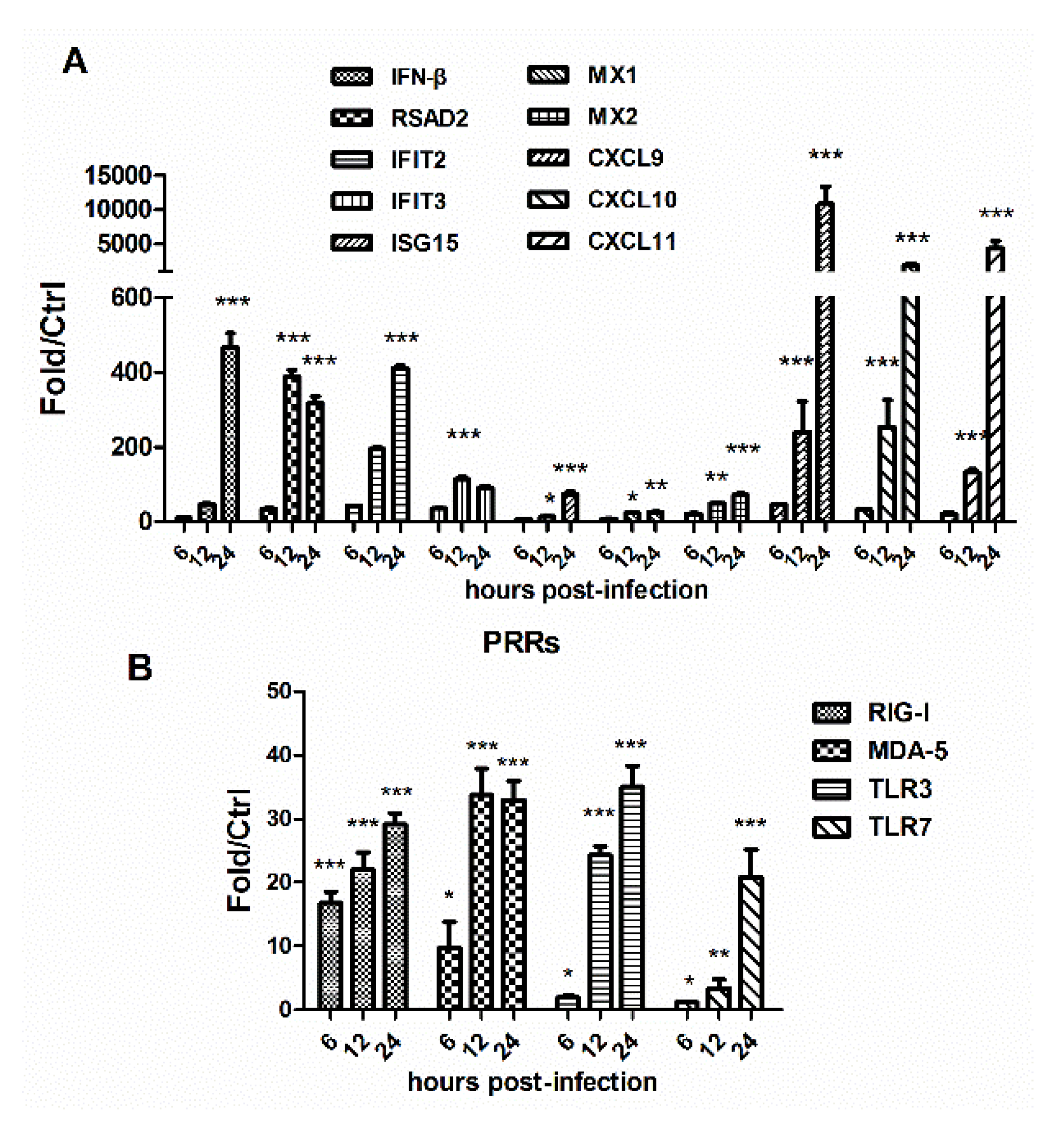
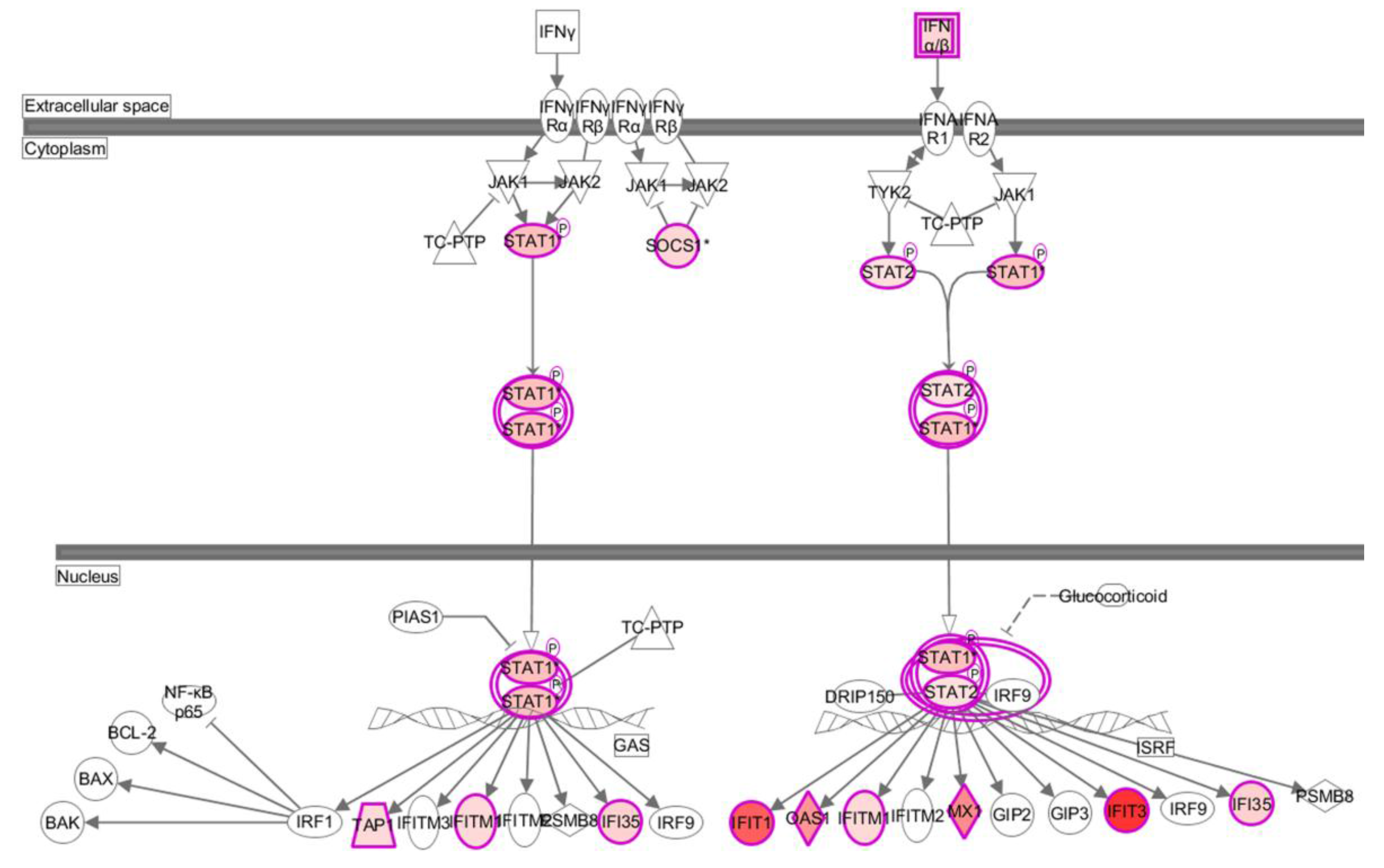
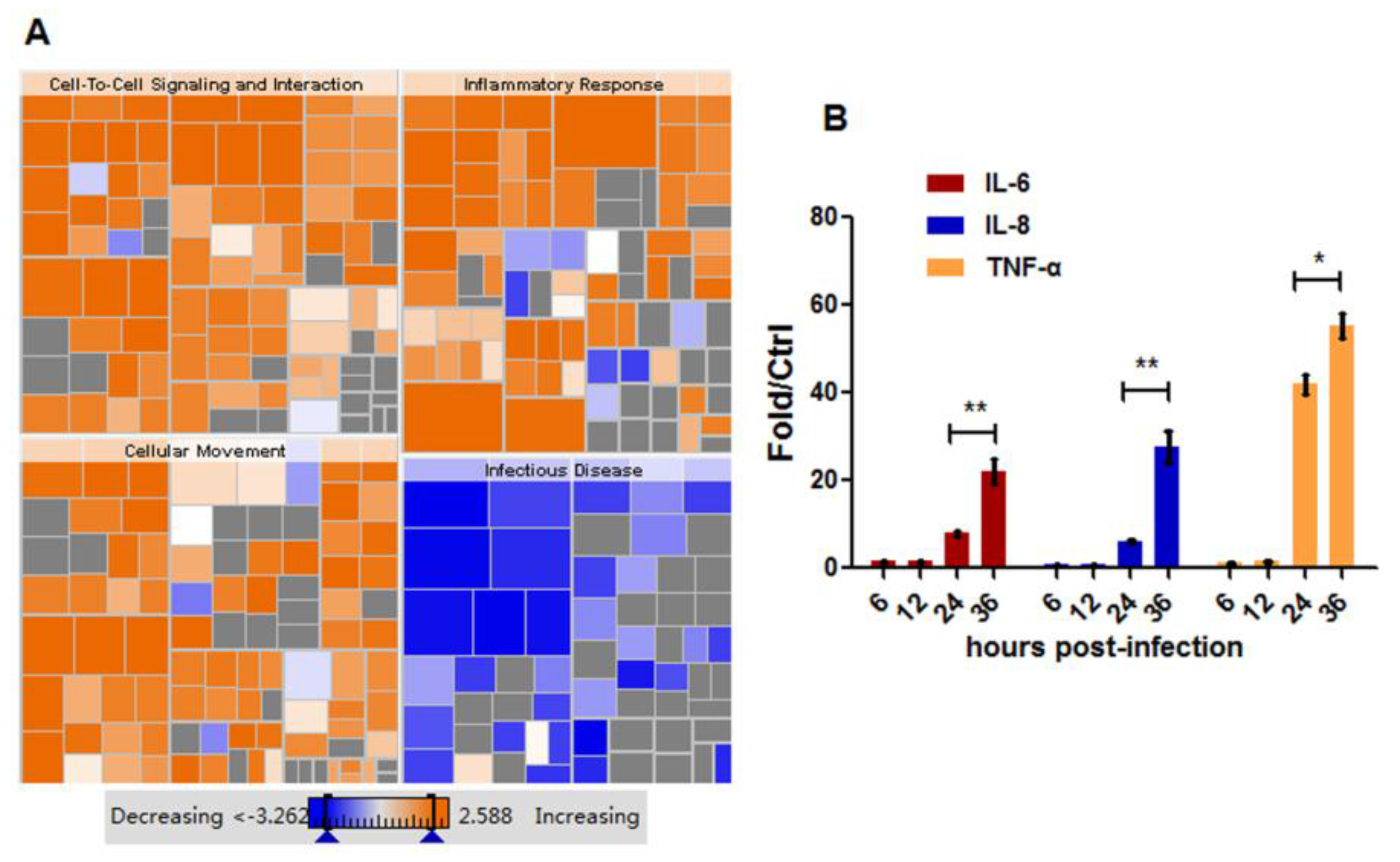
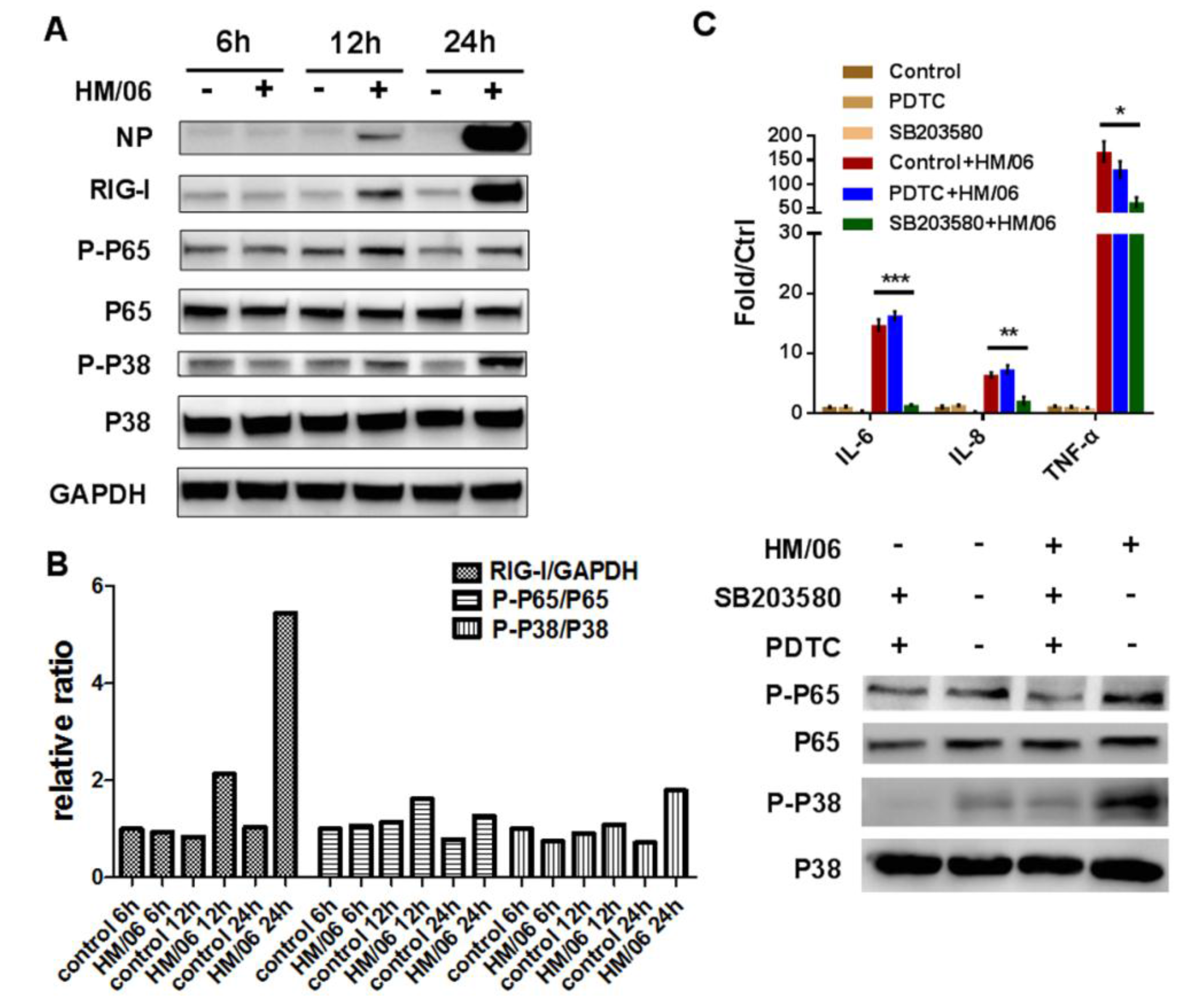
3.3. HM/06 Infection Triggered a Strong Innate Immune Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6 h | 12 h | 24 h | ||||||
|---|---|---|---|---|---|---|---|---|
| Gene Symbol | p-Value | Fold Change | Gene Symbol | p-Value | Fold Change | Gene Symbol | p-Value | Fold Change |
| FOSB | 9.52E-03 | 9.37 | MS4A6A | 9.80E-03 | 7.08 | RSAD2 | 8.20E-04 | 30.71 |
| LRTM2 | 2.20E-02 | 7.91 | OR5I1 | 1.05E-02 | 6.74 | IFIT2 | 5.18E-03 | 20.90 |
| FOS | 7.66E-04 | 5.64 | MPL | 1.90E-02 | 6.63 | IFIT3 | 4.96E-04 | 20.53 |
| MS4A6A | 7.86E-03 | 5.28 | OR4K17 | 3.52E-03 | 5.52 | CXCL10 | 1.47E-02 | 12.73 |
| ZIM3 | 3.65E-03 | 5.27 | RSAD2 | 2.70E-03 | 5.51 | OASL | 2.04E-03 | 11.12 |
| MYO1G | 2.01E-02 | 5.04 | CD34 | 2.00E-03 | 5.28 | MX2 | 2.62E-03 | 10.97 |
| OR4C46 | 3.37E-02 | 4.88 | MYO1G | 3.56E-02 | 4.39 | BATF2 | 8.10E-03 | 10.93 |
| TFF3 | 1.46E-02 | 4.51 | SLC6A20 | 1.69E-03 | 4.25 | CXCL11 | 1.21E-02 | 10.14 |
| LHX5 | 2.22E-02 | 4.48 | ZFP57 | 4.00E-02 | 4.23 | IFIT1 | 6.01E-04 | 9.99 |
| BEST3 | 1.22E-02 | 4.43 | OR8D1 | 1.57E-02 | 4.13 | DDX58 | 3.99E-04 | 9.55 |
| HTR3B | 1.12E-03 | 4.20 | IFIT3 | 3.71E-03 | 4.02 | OAS2 | 2.87E-03 | 9.41 |
| HGFAC | 1.55E-02 | 4.09 | TFF3 | 3.67E-02 | 3.92 | IFI44L | 3.79E-03 | 7.73 |
| FAM71B | 1.56E-02 | 4.02 | TTLL6 | 2.30E-02 | 3.9 | RTP4 | 3.04E-03 | 7.25 |
| AEBP1 | 3.07E-02 | 3.73 | RBFOX1 | 2.47E-03 | 3.87 | MX1 | 2.28E-03 | 7.13 |
| LBP | 1.13E-02 | 3.66 | ST6GALNAC1 | 1.79E-02 | 3.86 | FOSB | 3.60E-03 | 6.89 |
| ZG16 | 9.93E-03 | 3.66 | KSR1 | 1.83E-02 | 3.80 | OAS1 | 2.72E-03 | 6.56 |
| TAS2R46 | 1.72E-02 | 3.63 | SLC24A2 | 7.03E-03 | 3.76 | VPREB1 | 3.78E-02 | 5.91 |
| MRAP | 2.57E-02 | 3.60 | RFX4 | 1.22E-02 | 3.75 | BST2 | 7.65E-04 | 5.76 |
| MCHR2 | 3.16E-02 | 3.58 | OR7A10 | 1.76E-02 | 3.54 | SAMD9 | 7.71E-04 | 5.73 |
| MEP1A | 7.00E-03 | 3.32 | CDSN | 2.07E-03 | 3.46 | USP18 | 9.88E-03 | 5.53 |


3.4. HM/06 Infection Induces Extensive Inflammatory Response

| hpi | Gene Symbol | Fold Change | p-Value |
|---|---|---|---|
| 6 | MPL | 2.27 | 1.35E-02 |
| IL21 | 2.63 | 1.78E-02 | |
| TNFSF8 | 2.03 | 3.78E-02 | |
| LEPR | 2.54 | 4.29E-02 | |
| CCR4 | 2.93 | 4.76E-02 | |
| 12 | IL21 | 3.23 | 2.29E-04 |
| CXCL9 | 2.21 | 3.41E-03 | |
| IL17A | 2.39 | 1.58E-02 | |
| MPL | 6.63 | 1.90E-02 | |
| 24 | IL17A | 3.05 | 2.75E-03 |
| INHBC | 2.05 | 2.87E-03 | |
| IL12RB1 | 2.03 | 2.94E-03 | |
| CXCL9 | 5.41 | 1.07E-02 | |
| CCL5 | 3.63 | 1.27E-02 | |
| CXCL10 | 12.73 | 1.47E-02 | |
| CCL4 | 2.11 | 1.51E-02 | |
| CCR4 | 2.44 | 2.27E-02 | |
| IFNB1 | 2.74 | 2.35E-02 | |
| LEPR | 2.43 | 2.43E-02 | |
| TNFSF15 | 2.34 | 2.62E-02 | |
| IL10RA | 2.49 | 2.80E-02 | |
| IL29 | 3.17 | 2.84E-02 | |
| CD274 | 2.14 | 3.07E-02 | |
| CTF1 | 2.20 | 3.10E-02 | |
| CXCL11 | 5.32 | 3.27E-02 | |
| TNFSF10 | 2.39 | 3.28E-02 | |
| CCL11 | 2.93 | 3.91E-02 | |
| CSF3R | 2.08 | 4.33E-02 |

3.5. H5N1 Modified Internal Ca2+and EGTA Treatment Decreased Virus-Induced Apoptosis
| hpi | Gene Symbol | p-Value | Fold Change |
|---|---|---|---|
| 6 | VIP | 5.28E-03 | 2.59 |
| CHRNB1 | 1.67E-02 | 2.22 | |
| MCHR2 | 3.16E-02 | 3.58 | |
| HCRTR1 | 3.87E-02 | 0.49 | |
| MC2R | 2.87E-02 | 2.11 | |
| LEPR | 4.29E-02 | 2.54 | |
| 12 | HTR1A | 1.03E-03 | 2.95 |
| NTSR1 | 6.50E-03 | 3.22 | |
| 24 | HTR1A | 1.86E-03 | 2.26 |
| GHRHR | 2.60E-03 | 2.13 | |
| CHRNG | 2.78E-03 | 2.36 | |
| GRM7 | 4.34E-03 | 2.01 | |
| SSTR2 | 5.44E-03 | 2.48 | |
| CGA | 1.07E-02 | 2.17 | |
| P2RY13 | 1.09E-02 | 4.50 | |
| OPRM1 | 1.11E-02 | 2.17 | |
| GABRA1 | 1.25E-02 | 2.33 | |
| MC3R | 1.38E-02 | 2.96 | |
| CNR2 | 1.76E-02 | 2.65 | |
| LEPR | 2.43E-02 | 2.43 | |
| CCKAR | 2.77E-02 | 2.56 | |
| HRH2 | 3.07E-02 | 2.12 | |
| MCHR2 | 3.34E-02 | 3.04 | |
| MC2R | 4.00E-02 | 2.46 | |
| HTR1B | 4.30E-02 | 2.62 | |
| NPB | 4.57E-02 | 2.28 | |
| MLNR | 4.77E-02 | 2.43 | |
| TAAR8 | 4.98E-02 | 2.00 |

4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fujimoto, S.; Kobayashi, M.; Uemura, O.; Iwasa, M.; Ando, T.; Katoh, T.; Nakamura, C.; Maki, N.; Togari, H.; Wada, Y. PCR on cerebrospinal fluid to show influenza-associated acute encephalopathy or encephalitis. Lancet 1998, 352, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Watanabe, S.; Ito, T.; Goto, H.; Wells, K.; McGregor, M.; Cooley, A.J.; Kawaoka, Y. Biological heterogeneity, including systemic replication in mice, of H5N1 influenza A virus isolates from humans in Hong Kong. J. Virol. 1999, 73, 3184–3189. [Google Scholar] [PubMed]
- Ito, Y.; Ichiyama, T.; Kimura, H.; Shibata, M.; Ishiwada, N.; Kuroki, H.; Furukawa, S.; Morishima, T. Detection of influenza virus RNA by reverse transcription-PCR and proinflammatory cytokines in influenza-virus-associated encephalopathy. J. Med. Virol. 1999, 58, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Itoh, M.; Mizuguchi, M.; Ozawa, H.; Okazaki, E.; Kobayashi, Y.; Takahashi, M.; Ohtani, K.; Ogawa, A.; Narita, M.; et al. Apoptosis and microglial activation in influenza encephalopathy. Acta Neuropathol. 2003, 105, 233–239. [Google Scholar]
- Shinjoh, M.; Yoshikawa, T.; Li, Y.; Shiraishi, K.; Ueki, H.; Nerome, K. Prophylaxis and treatment of influenza encephalitis in an experimental mouse model. J. Med. Virol. 2002, 67, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Shinya, K.; Silvano, F.D.; Morita, T.; Shimada, A.; Nakajima, M.; Ito, T.; Otsuki, K.; Umemura, T. Encephalitis in mice inoculated intranasally with an influenza virus strain originated from a water bird. J. Vet. Med. Sci. 1998, 60, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Gambotto, A.; Barratt-Boyes, S.M.; de Jong, M.D.; Neumann, G.; Kawaoka, Y. Human infection with highly pathogenic H5N1 influenza virus. Lancet 2008, 371, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.D.; Bach, V.C.; Phan, T.Q.; Vo, M.H.; Tran, T.T.; Nguyen, B.H.; Beld, M.; Le, T.P.; Truong, H.K.; Nguyen, V.V.; et al. Fatal avian influenza A (H5N1) in a child presenting with diarrhea followed by coma. N. Engl. J. Med. 2005, 352, 686–691. [Google Scholar]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.; McClaren, J.; Pani, A.K.; Smeyne, M.; Korff, A.; Webster, R.; Smeyne, R.J. Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J. Neurosci. 2012, 32, 1545–1559. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, J.; Li, W.; Xin, G.; Su, Y.; Gao, Y.; Zhang, H.; Lin, G.; Jiao, X.; Li, K. Apoptosis and proinflammatory cytokine responses of primary mouse microglia and astrocytes induced by human H1N1 and avian H5N1 influenza viruses. Cell Mol. Immunol. 2008, 5, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.P.; Lee, S.M.; Cheung, T.K.; Nicholls, J.M.; Peiris, J.S.; Ip, N.Y. Avian influenza H5N1 virus induces cytopathy and proinflammatory cytokine responses in human astrocytic and neuronal cell lines. Neuroscience 2010, 168, 613–623. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar]
- To, K.F.; Chan, P.K.; Chan, K.F.; Lee, W.K.; Lam, W.Y.; Wong, K.F.; Tang, N.L.; Tsang, D.N.; Sung, R.Y.; Buckley, T.A.; et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J. Med. Virol. 2001, 63, 242–246. [Google Scholar]
- Zou, W.; Yu, Z.; Zhou, H.; Tu, J.; Jin, M. Genetic characterization of an H5N1 avian influenza virus with neurovirulence in ducks. Virus Genes 2009, 38, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.J.; Winsor, G.L.; Chan, C.; Richard, N.; Laird, M.R.; Barsky, A.; Gardy, J.L.; Roche, F.M.; Chan, T.H.; Shah, N.; et al. InnateDB: Facilitating systems-level analyses of the mammalian innate immune response. Mol. Syst. Biol. 2008, 4. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science 1990, 247, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Goodman, A.G.; Korth, M.J.; Katze, M.G. Hijacking of the host-cell response and translational control during influenza virus infection. Virus Res. 2006, 119, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; de Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar]
- Bonavia, R.; Bajetto, A.; Barbero, S.; Albini, A.; Noonan, D.M.; Schettini, G. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem. Biophys. Res. Commun. 2001, 288, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Daidoji, T.; Du, A.; Yang, C.S.; Ibrahim, M.S.; Ikuta, K.; Nakaya, T. Highly pathogenic H5N1 avian influenza virus induces extracellular Ca2+ influx, leading to apoptosis in avian cells. J. Virol. 2010, 84, 3068–3078. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nikrad, M.P.; Travanty, E.A.; Zhou, B.; Phang, T.; Gao, B.; Alford, T.; Ito, Y.; Nahreini, P.; Hartshorn, K.; et al. Innate immune response of human alveolar macrophages during influenza A infection. PLoS ONE 2012, 7, e29879. [Google Scholar]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 2011, 162, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.C.; Cresswell, P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2001, 98, 15125–15130. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hinson, E.R.; Cresswell, P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2007, 2, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Olfat, F.; Phoon, M.C.; Hsu, J.P.; Howe, J.L.; Seet, J.E.; Chin, K.C.; Chow, V.T. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J. Gen. Virol. 2012, 93, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 2007, 81, 11246–11255. [Google Scholar] [CrossRef] [PubMed]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 2011, 31, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.A.; Gorna, M.W.; Baumann, C.L.; Burkard, T.R.; Stefanovic, A.; Burckstummer, T.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5'-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar]
- Jiang, D.; Guo, H.; Xu, C.; Chang, J.; Gu, B.; Wang, L.; Block, T.M.; Guo, J.T. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 2008, 82, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Shi, P.Y.; Block, T.M.; et al. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, J.; Yang, B.; Zheng, N.; Qin, M.; Ji, Y.; Lin, G.; Tian, L.; Wu, X.; Wu, L.; Sun, B. Guanylate binding protein 4 negatively regulates virus-induced type I IFN and antiviral response by targeting IFN regulatory factor 7. J. Immunol. 2011, 187, 6456–6462. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, C.; Xue, P.; Zhong, B.; Mao, A.P.; Ran, Y.; Chen, H.; Wang, Y.Y.; Yang, F.; Shu, H.B. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. USA 2009, 106, 7945–7950. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Hwang, S.Y.; Imaizumi, T.; Yoo, J.Y. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J. Virol. 2008, 82, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.; Poon, L.L.; Lau, A.S.; Luk, W.; Lau, Y.L.; Shortridge, K.F.; Gordon, S.; Guan, Y.; Peiris, J.S. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: A mechanism for the unusual severity of human disease? Lancet 2002, 360, 1831–1837. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.A.; Plowden, J.K.; Garcia-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008, 4, e1000115. [Google Scholar] [CrossRef] [PubMed]
- Baskin, C.R.; Bielefeldt-Ohmann, H.; Tumpey, T.M.; Sabourin, P.J.; Long, J.P.; Garcia-Sastre, A.; Tolnay, A.E.; Albrecht, R.; Pyles, J.A.; Olson, P.H.; et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. USA 2009, 106, 3455–3460. [Google Scholar]
- Cameron, C.M.; Cameron, M.J.; Bermejo-Martin, J.F.; Ran, L.; Xu, L.; Turner, P.V.; Ran, R.; Danesh, A.; Fang, Y.; Chan, P.K.; et al. Gene expression analysis of host innate immune responses during Lethal H5N1 infection in ferrets. J. Virol. 2008, 82, 11308–11317. [Google Scholar]
- Hui, K.P.; Lee, S.M.; Cheung, C.Y.; Ng, I.H.; Poon, L.L.; Guan, Y.; Ip, N.Y.; Lau, A.S.; Peiris, J.S. Induction of proinflammatory cytokines in primary human macrophages by influenza A virus (H5N1) is selectively regulated by IFN regulatory factor 3 and p38 MAPK. J. Immunol. 2009, 182, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Deitmer, J.W.; Verkhratsky, A.J.; Lohr, C. Calcium signaling in glial cells. Cell Calcium 1998, 24, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Orkand, R.K.; Kettenmann, H. Glial calcium: homeostasis and signaling function. Physiol. Rev. 1998, 78, 99–141. [Google Scholar] [PubMed]
- Haydon, P.G.; Carmignoto, G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- Haydon, P.G. GLIA: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Park, C.H.; Ishinaka, M.; Takada, A.; Kida, H.; Kimura, T.; Ochiai, K.; Umemura, T. The invasion routes of neurovirulent A/Hong Kong/483/97 (H5N1) influenza virus into the central nervous system after respiratory infection in mice. Arch. Virol. 2002, 147, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Shinya, K.; Shimada, A.; Ito, T.; Otsuki, K.; Morita, T.; Tanaka, H.; Takada, A.; Kida, H.; Umemura, T. Avian influenza virus intranasally inoculated infects the central nervous system of mice through the general visceral afferent nerve. Arch. Virol. 2000, 145, 187–195. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Wang, R.; Zhang, J.; Sun, X.; Zou, Z.; Wang, S.; Jin, M. Insights into Human Astrocyte Response to H5N1 Infection by Microarray Analysis. Viruses 2015, 7, 2618-2640. https://doi.org/10.3390/v7052618
Lin X, Wang R, Zhang J, Sun X, Zou Z, Wang S, Jin M. Insights into Human Astrocyte Response to H5N1 Infection by Microarray Analysis. Viruses. 2015; 7(5):2618-2640. https://doi.org/10.3390/v7052618
Chicago/Turabian StyleLin, Xian, Ruifang Wang, Jun Zhang, Xin Sun, Zhong Zou, Shengyu Wang, and Meilin Jin. 2015. "Insights into Human Astrocyte Response to H5N1 Infection by Microarray Analysis" Viruses 7, no. 5: 2618-2640. https://doi.org/10.3390/v7052618