
Using the Hepatitis C Virus RNA-Dependent RNA Polymerase as a Model to Understand Viral Polymerase Structure, Function and Dynamics

Abstract
:1. Introduction
- (1)
- Recognition of the nucleic acid binding site
- (2)
- Coordination of the chemical steps of nucleic acid synthesis
- (3)
- Conformational rearrangement to allow for processive elongation
- (3)
- Termination of replication at the end of the genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Material | Baltimore Classification | Polymerase Classes | Examples |
|---|---|---|---|
| DNA | ssDNA viruses | DNA dependent DNA polymerases | Human parvovirus B19 |
| dsDNA viruses | DNA dependent RNA polymerases | Bacteriophage φ29 | |
| RNA | (+) ssRNA viruses | RNA dependent RNA polymerases | HCV, PV, West Nile virus |
| (−) ssRNA viruses | Influenza | ||
| dsRNA | Bacteriophage φ6 | ||
| RNA/ | ssRNA-rt viruses | RNA dependent DNA polymerases | Retrovirus |
| DNA | dsDNA-rt viruses | Hepatitis B |
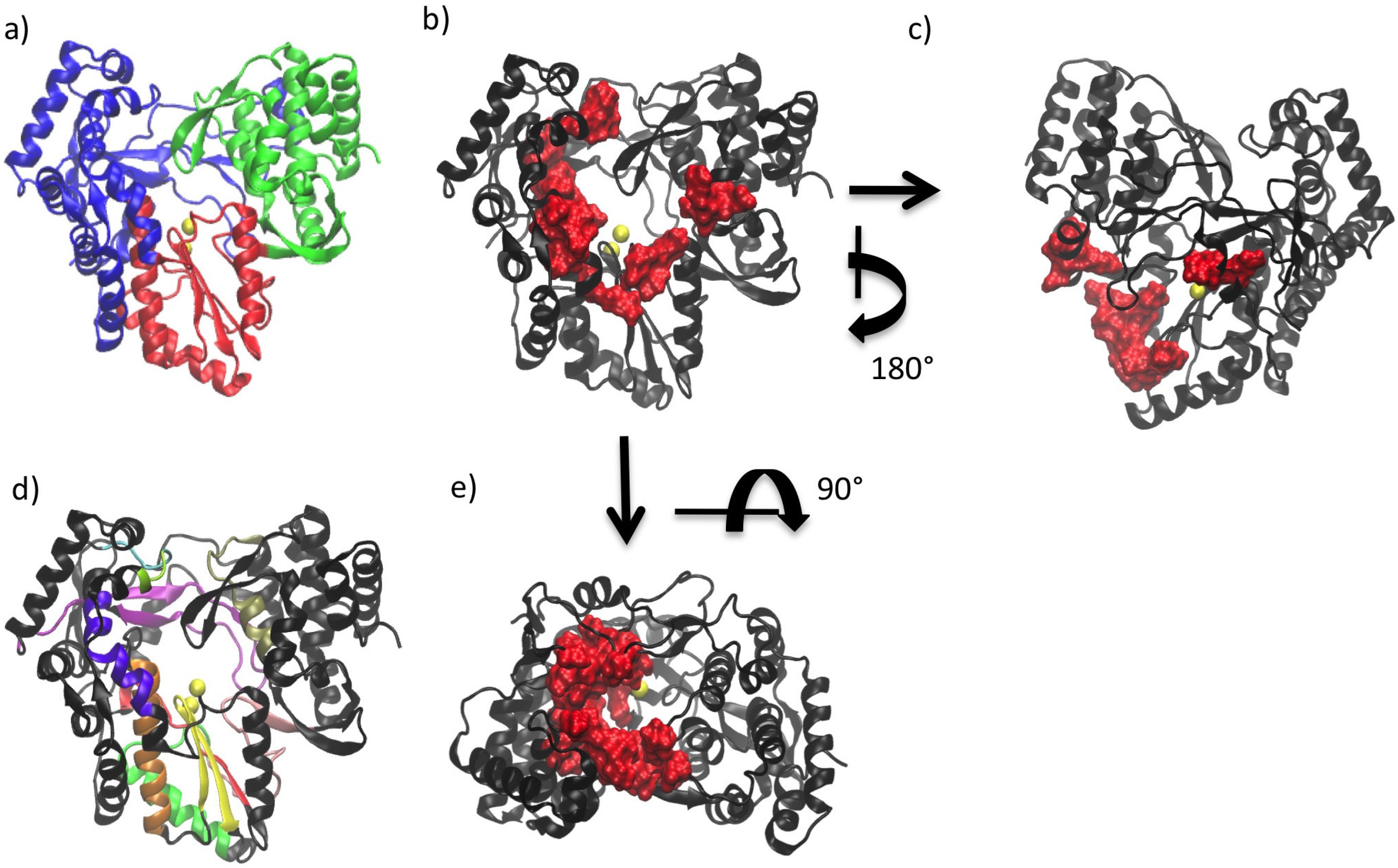
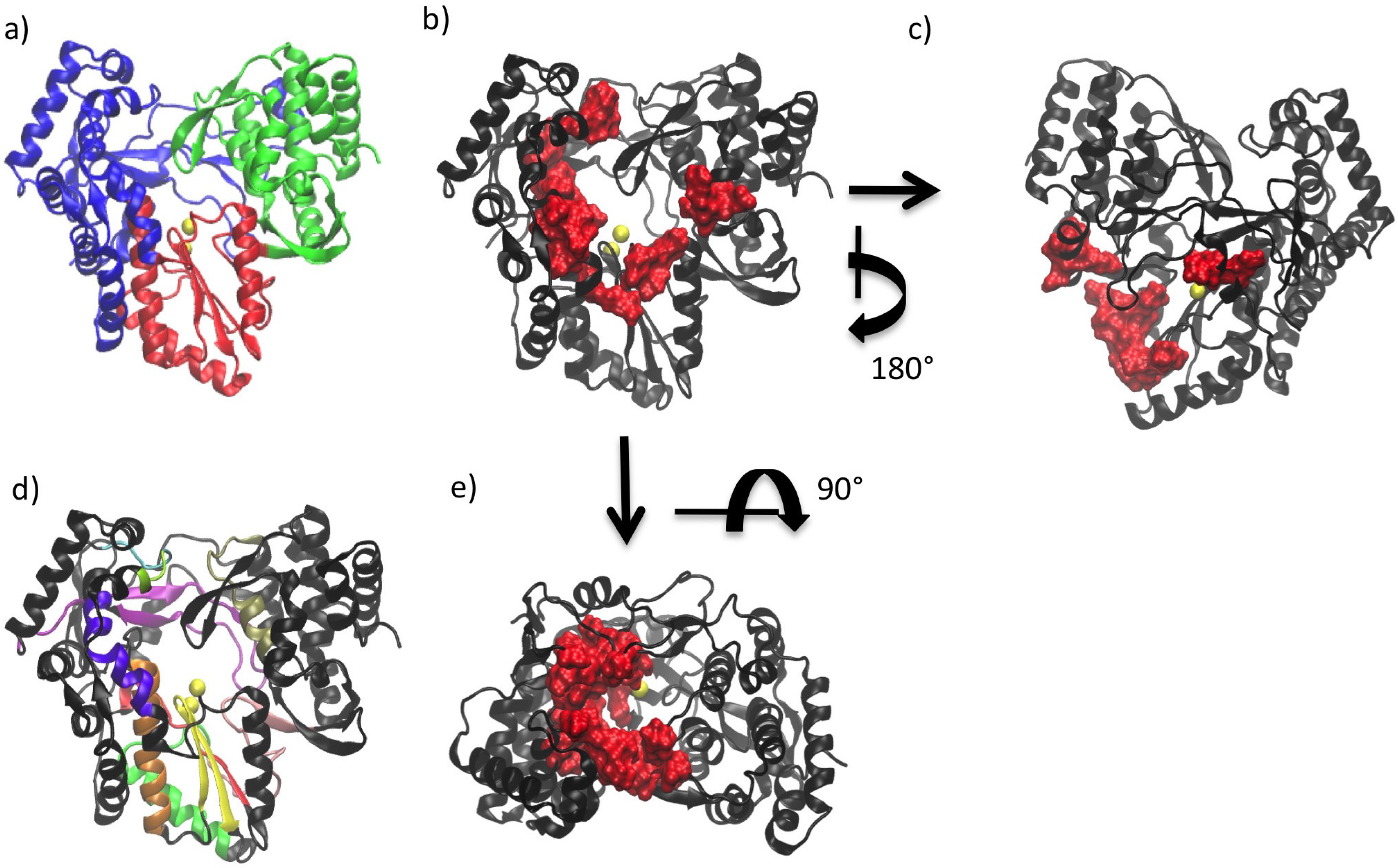
2. General Structural Features of Viral Polymerases
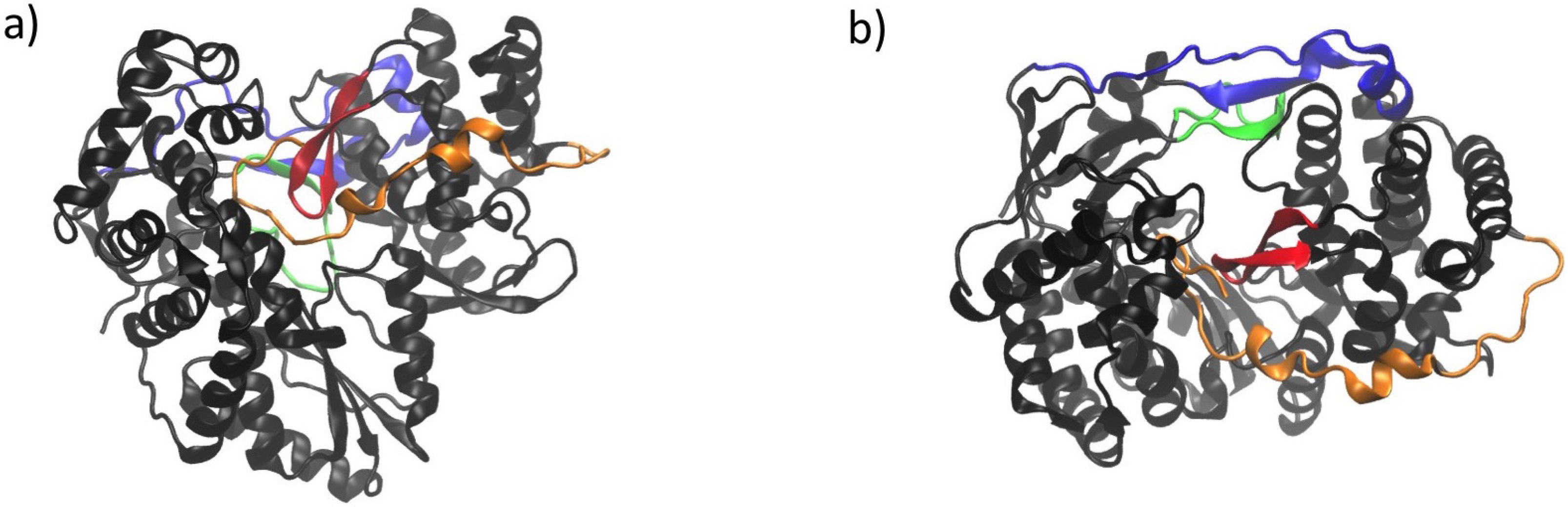
3. Conserved Structural Motifs of Viral Polymerases
| Conserved Elements | Role | Location | Residues | |||
|---|---|---|---|---|---|---|
| HCV | PV | FMDV | ||||
| Motifs | A | Coordinates Magnesium and selects type of nucleic acid (RNA vs. DNA) | Palm | 216–227 | 229–240 | 236–247 |
| B | Determines nucleotide choice (rNTP or dNTP) | Palm | 287–306 | 293–312 | 303–322 | |
| C | Coordinates Magnesium | Palm | 312–325 | 322–335 | 332–345 | |
| D | Helps accommodate active site NTPs | Palm | 332–353 | 338–362 | 348–373 | |
| E | Maintains rigidity of secondary structure that is required for relative positioning of thumb and palm domains | Palm | 354–372 | 363–380 | 374–392 | |
| F | Binds incoming NTPs and RNA | Fingers | 132–162 | 153–178 | 158–183 | |
| G | Binds primer and template | Fingers | 95–99 | 113–120 | 114–121 | |
| Functional regions | I | Binds template | Fingers | 91–94 | 107–112 | 108–113 |
| II | Binds template | Fingers | 168–183 | 184–200 | 189–205 | |
| III | Binds nascent RNA duplex | Thumb | 401–414 | 405–420 | 416–430 | |
| Virus family | Representative Species | |
|---|---|---|
| (+) ssRNA | Picornaviradae | Poliovirus (PV) |
| Human rhinovirus (HRV) | ||
| Foot-and-mouth-disease virus (FMDV) | ||
| Coxsackie viruses (CV) | ||
| Hepatitis A virus (HAV) | ||
| Caliciviridae | Rabbit hemorrhagic disease virus (RHDV) | |
| Norwalk virus (NV) | ||
| Sapporo virus | ||
| Togaviridae | Sindbis virus | |
| Flaviviridae | West Nile virus (WNV) | |
| Yellow fever virus | ||
| Dengue virus (DENV) | ||
| Japanese encephalitis disease virus (JEV) | ||
| Hepatitis C virus (HCV) | ||
| Bovine viral diarrhea virus (BVDV) | ||
| (−) ssRNA | Orthomyxoviridae | Influenza virus |
| Paramyxoviridae | Measles and mumps viruses | |
| Bunyaviridae | Hantavirus | |
| Rhabdoviridae | Rabies virus | |
| Filoviridae | Ebola and Marburg virus | |
| Bornaviridae | Borna disease virus | |
| dsRNA | Cystoviridae | Bacteriophage ϕ6 |
| Reoviridae | Reovirus | |
| Birnaviridae | Fish infectious pancreatic necrosis virus (IPNV) | |
| Infectious bursal disease virus (IBDV) |
4. Structural Features of RdRps
| Virus Family | Genus | Species |
|---|---|---|
| Flaviviridae | Flaviviruses | West Nile virus |
| Yellow fever virus | ||
| Dengue virus | ||
| Japanese encephalitis disease virus | ||
| Hepaciviruses | Hepatitis C virus (HCV) | |
| Pestiviruses | Bovine viral diarrhea virus (BVDV) |
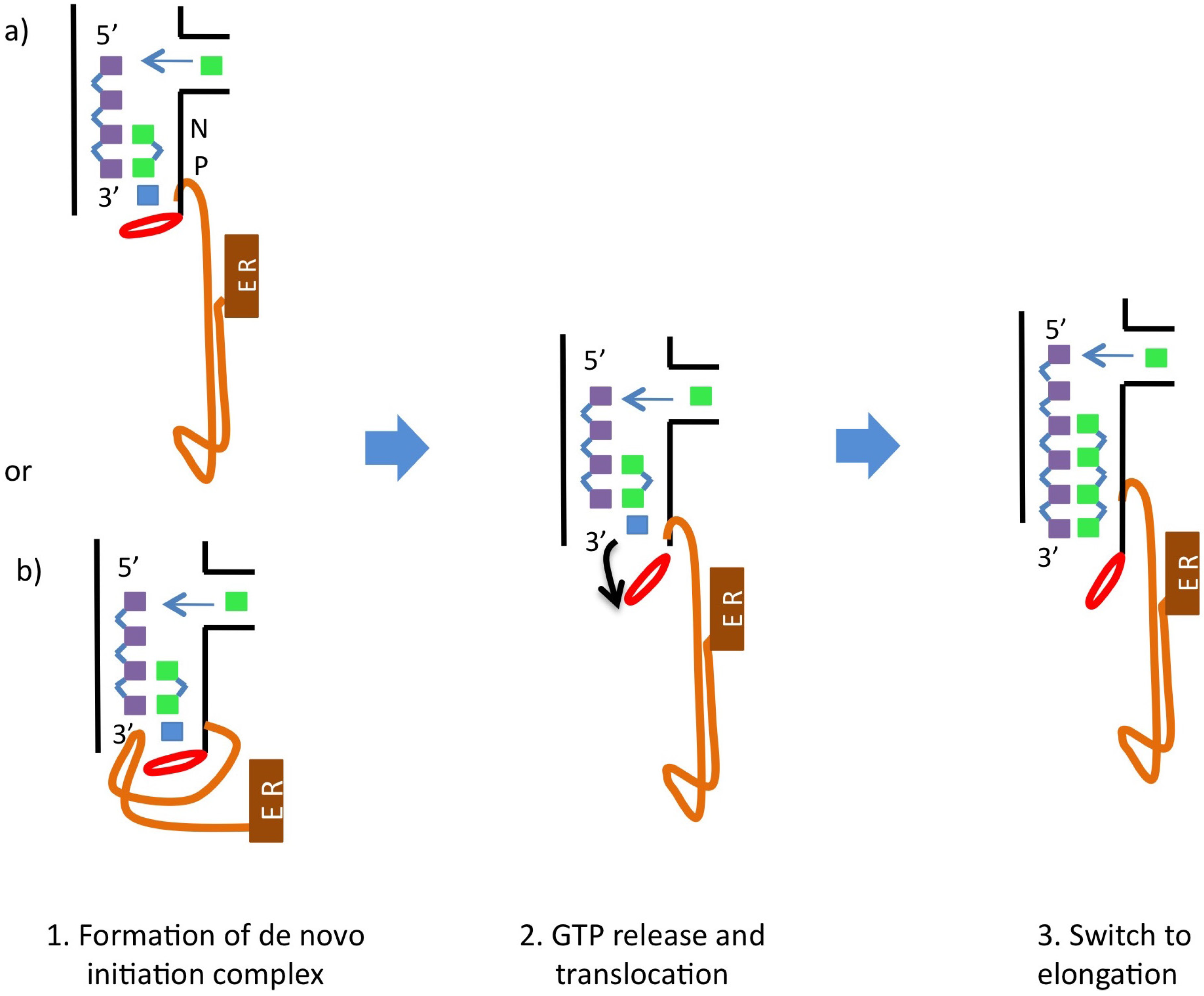
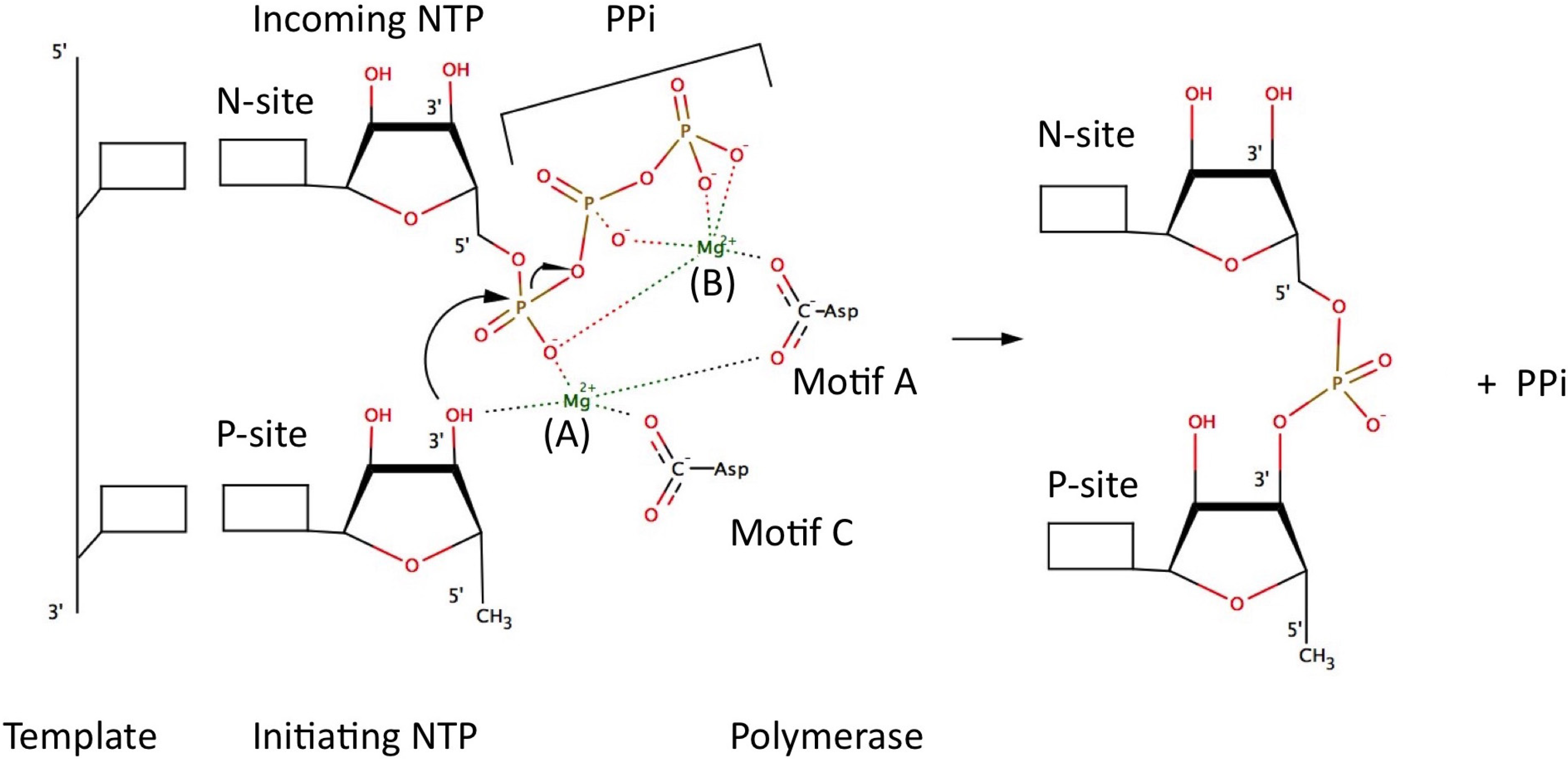
5. Catalytic Mechanism and Polymerase Reaction Steps


- (1)
- incorporation of the incoming NTP into the growing daughter strand by formation of the phosphodiester bond
- (2)
- release of pyrophosphate
- (3)
- translocation along the template.

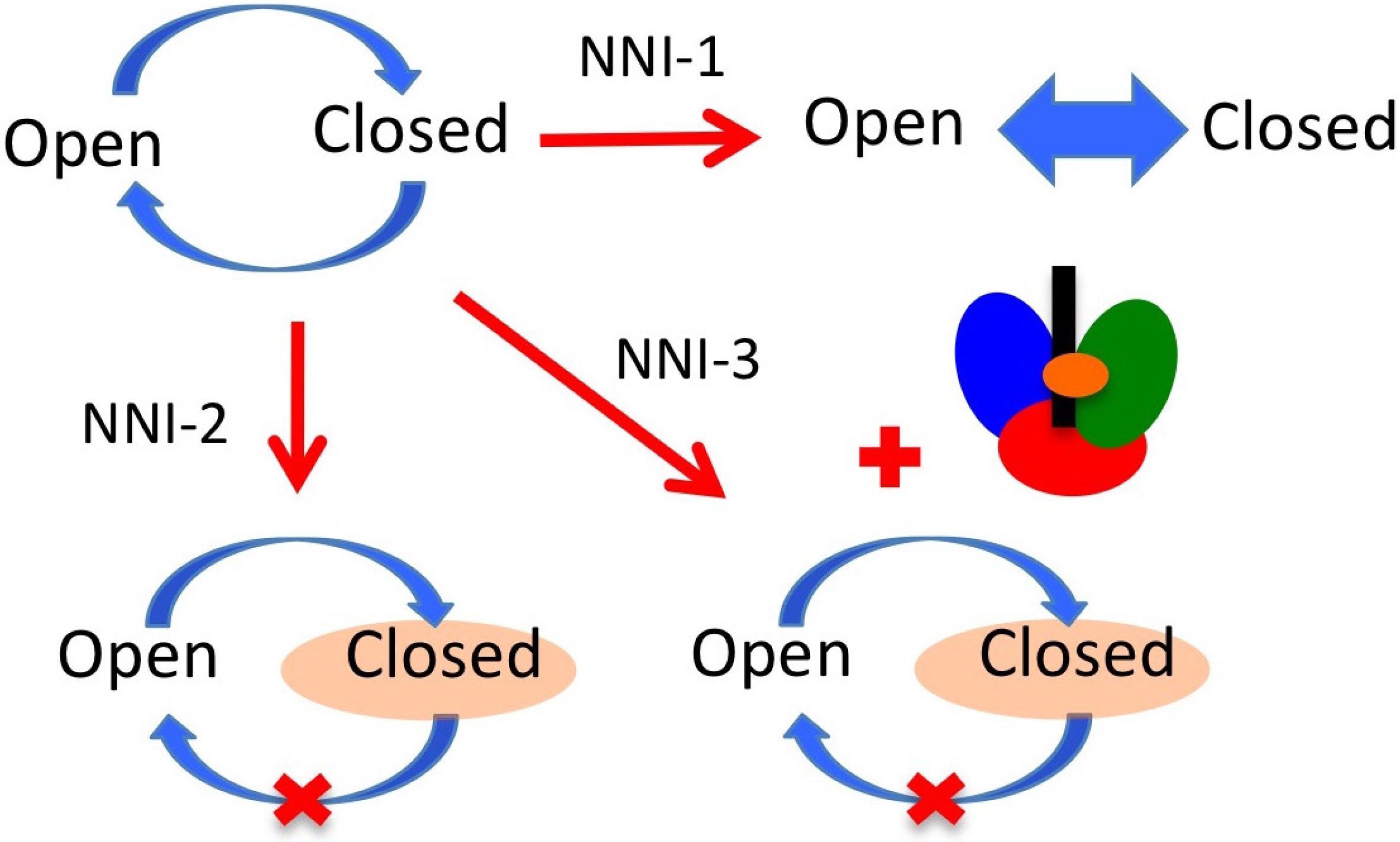
6. NS5B Conformational Changes during the Replication Cycle
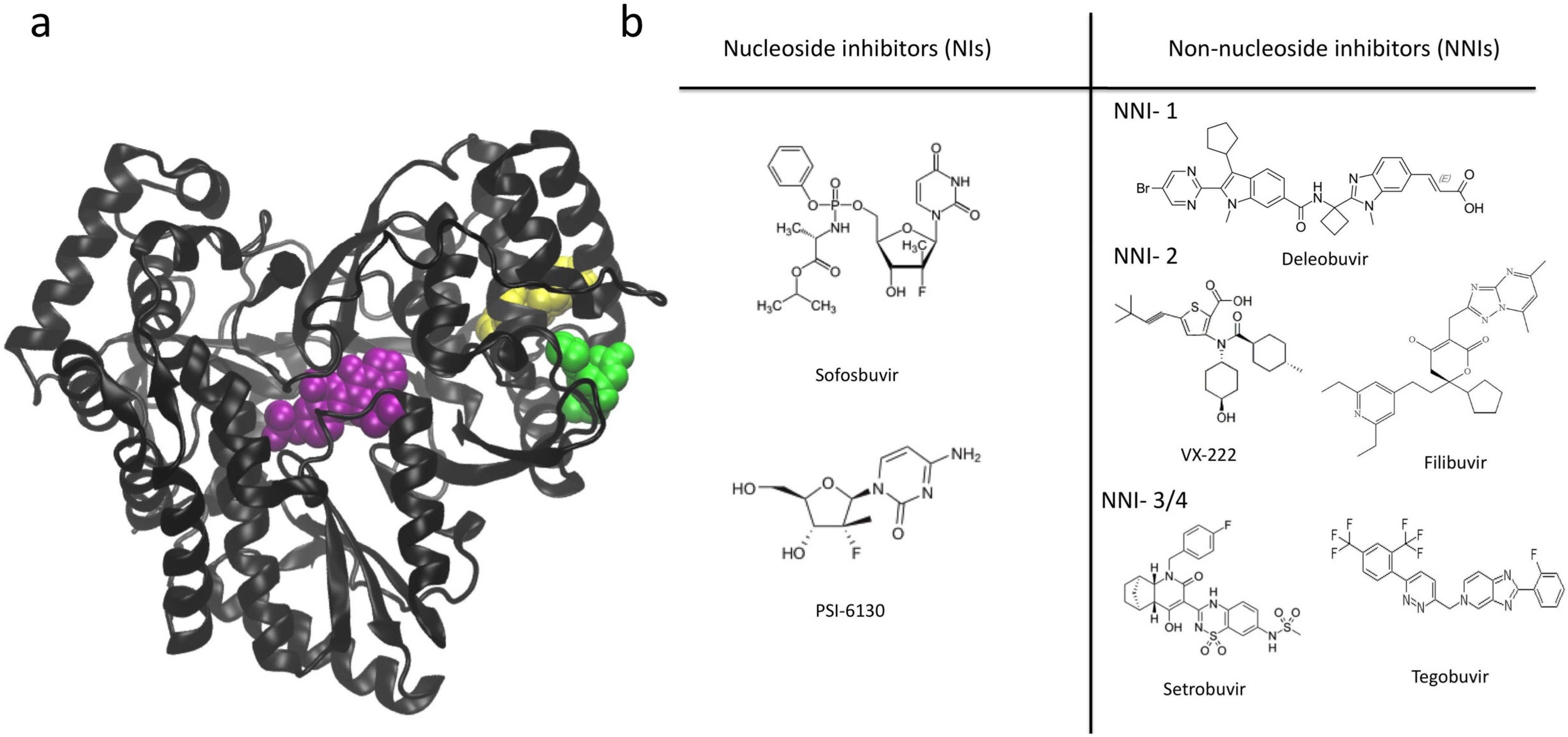
7. NS5B Inhibitors and Mechanisms of Action


8. Summary
Conflicts of Interest
References
- Choi, K.H. Viral polymerases. In Viral Molecular Machines; Springer Science: New York, NY, USA, 2012. [Google Scholar]
- Ortin, J.; Parra, F. Structure and function of RNA replication. Annu. Rev. Microbiol. 2006, 60, 305–326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.-Y.; Li, H.; et al. Structure and function of flavivirus ns5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chung, S.; Miller, M.; Grice, S.F.J.L.; Wlodawer, A. Crystal structures of the reverse transcriptase-associated ribonuclease h domain of xenotropic murine leukemia-virus related virus. J. Struct. Biol. 2012, 177, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Knopf, C. Evolution of viral DNA-dependent DNA polymerases. Virus Genes 1998, 16, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [PubMed]
- Shatskaya, G.S. Structural organization of viral RNA-dependent RNA polymerases. Biochemistry 2013, 78, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Ollis, D.L.; Brick, P.; Hamlin, R.; Xuong, N.G.; Steitz, T.A. Structure of large fragment of Escherichia coli DNA polymerase i complexed with dtmp. Nature 1985, 313, 762–766. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M. RNA synthetic mechanisms employed by diverse families of RNA viruses. WIREs RNA 2013, 4, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Orta, C.; Verdaguer, N. RNA virus polymerases. In Viral Genome Replication; Cameron, C., Gotte, M., Raney, K.D., Eds.; Springer Science: New York, NY, USA, 2009. [Google Scholar]
- Gao, G.; Orlova, M.; Georgiadis, M.M.; Hendrickson, W.A.; Goff, S.P. Conferring RNA polymerase activity to a DNA polymerase: A single residue in reverse transcriptase controls substrate selection. Proc. Natl. Acad. Sci. USA 1997, 94, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Moustafa, I.M.; Arnold, J.J. Dynamics: The missing link between structure and function of the viral RNA-dependent RNA polymerase? Curr. Opin. Struct. Biol. 2009, 19, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.K.-S.; Arnold, J.J.; Cameron, C.E. Structure and Function Relationships Ammong RNA-Dependent RNA Polymerases; Springer-Verlag: Berlin, Germany; Heidelberg, Germany, 2008; Volume 320. [Google Scholar]
- Subissi, L.; Decroly, E.; Selisko, B.; Canard, B.; Imbert1, I. A closed-handed affair: Positive-strand RNA virus polymerases. Future Virol. 2014, 9, 769–784. [Google Scholar] [CrossRef]
- Moustafa, I.M.; Shen, H.; Morton, B.; Colina, C.M.; Cameron, C.E. Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity. J. Mol. Biol. 2011, 410, 159–181. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Thorpe, I.F. Thumb inhibitor binding eliminates functionally important dynamics in the hepatitis c virus RNA polymerase. Proteins Struct. Funct. Bioinform. 2013, 81, 40–52. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: http://www.Ictvonline.Org (accessed on February 15,2015).
- Gong, J.; Fang, H.; Li, M.; Liu, Y.; Yang, K.; Xu, W. Potential targets and their relevant inhibitors in anti-influenza fields. Curr. Med. Chem. 2009, 16, 3716–3739. [Google Scholar] [CrossRef] [PubMed]
- Malet, H.; Masse, N.; Selisko, B.; Romette, J.L.; Alvarez, K.; Guillemot, J.C.; Tolou, H.; Yap, T.L.; Vasudevan, S.; Lescar, J.; et al. The flavivirus polymerase as a target for drug discovery. Antivir. Res. 2008, 2008, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Ilnytska, O.; Belov, G. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, A.J. Hepatitis viruses. In Medical Microbiology; Baron, S., Ed.; The University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Hansen, J.L.; Long, A.M.; Schultz, S.C. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure 1997, 5, 1109–1122. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Tellinghuisen, T.L. Hepatitis C virus genome replication. In Viral Genome Replication; Cameron, C., Gotte, M., Raney, K.D., Eds.; Springer Science: New York, NY, USA, 2009. [Google Scholar]
- Astier-Manifacier, S.; Cornuet, P. RNA-dependent RNA polymerase in chinese cabbage. Biochim. Biophys. Acta 1971, 232, 484–493. [Google Scholar] [CrossRef]
- Boege, F.; Heinz, L.S. RNA-dependent RNA polymerase from healthy tomato leaf tissue. FEBS Lett. 1980, 121, 91–96. [Google Scholar] [CrossRef]
- Cogoni., C.; Macino, G. Gene silencing in neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature 1999, 399, 166–169. [Google Scholar] [PubMed]
- Smardon, A.; Spoerke, J.M.; Stacey, S.C.; Klein, M.E.; Mackin, N.; Maine, E.M. Ego-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in c. Elegans. Curr. Biol. 2000, 10, 169–178. [Google Scholar] [CrossRef]
- Maida, Y.; Masutomi, K. RNA-dependent RNA polymerases in RNA silencing. Biol. Chem. 2011, 392, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Rohayem, J.; Robel, I.; Jager, K.; Scheffler, U.; Rudolph, W. Protein-primed and de novo initiation of RNA synthesis by norovirus 3dpol. J. Virol. 2006, 80, 7060–7069. [Google Scholar] [CrossRef] [PubMed]
- Ranjith-Kumar, C.T.; Kao, C.C. Recombinant viral rdrps can initiate RNA synthesis from circular templates. RNA 2006, 12, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Hamatake, R.K.; Mathis, D.M.; Racela, J.; Rigat, K.L.; Lemm, J.; Colonno, R.J. De novo initiation of RNA synthesis by the RNA-dependent RNA polymerase (ns5b) of hepatitis C virus. J. Virol. 2000, 74, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.C.; Vecchio, A.M.D.; Zhong, W. De novo initiation of RNA synthesis by a recombinant flaviviridae RNA-dependent RNA polymerase. Virology 1999, 253, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Harrus, D. Further insights into the roles of GTP and the C terminus of the hepatitis C virus polymerase in the initiation of RNA synthesis. J. Biol. Chem. 2010, 285, 32906–32918. [Google Scholar] [CrossRef] [PubMed]
- D’Abramo, C.M.; Deval, J.; Cameron, C.E.; Cellai, L.; Gotte, M. Control of template positioning during de novo initiation of RNA synthesis by the bovine viral diarrhea virus NS5B polymerase. J. Biol. Chem. 2006, 281, 24991–24998. [Google Scholar] [CrossRef] [PubMed]
- Bressanelli, S. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 2002, 76, 3482–3492. [Google Scholar] [CrossRef] [PubMed]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D., III; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Structural basis for RNA replication by the hepatitis C virus polymerase. Science 2015, 347, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.A.; Makeyev, E.V.; Bamford, D.H. Initation of viral RNA-dependent RNA polymerization. J. Gen. Virol. 2004, 85, 1077–1093. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T. A mechanism for all polymerases. Nature 1998, 391, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V. Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer-Verlag: Berlin, Germany; Heidelberg, Germany, 2013; Volume 369. [Google Scholar]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Orta, C.; Arias, A.; Escarmi, C.; Verdaguer, N. A comparison of viral RNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2006, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.; Quinckert, D.; Bochkarova, O.; Klein, R.; Kezmic, N.; Bartenschalager, R.; Lohmann, V. Identification of determinants involved in initatiation of hepatitis c virus RNA synthesis by using intergenotipic chimeras. J. Virol. 2007, 81, 5270–5283. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Larson, G.; Hong, J.Z. Selection of 3′ template bases and initatiting nucleotides by hepatitis c virus RNA by and ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 2002, 109, 941–946. [Google Scholar]
- Biswal, B.K.; Cherney, M.M.; Wang, M.; Chan, L.; Yannopoulos, C.G.; Bilimoria, D.; Nicolas, O.; Bedard, J.; James, M.N. Crystal structures of the RNA-dependent RNA polymerase genotype 2A of hepatitis C virus reveal two conformations and suggest mechanisms of inhibition by non-nucleoside inhibitors. J. Biol. Chem. 2005, 280, 18202–18210. [Google Scholar] [CrossRef] [PubMed]
- Mosley, R.T. Structure of hepatitis C virus polymerase in complex with primer- template RNA. J. Virol. 2012, 86, 6503–6511. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.C.; Brown, J.A.; Thorpe, I.F. Allosteric inhibitors have distinct effects, but also common modes of action, in the hcv polymerase. Biophys. J. 2015, 108, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Caillet-Saguy, C.; Lim, S.P.; Shi, P.-Y.; Lescar, J.; Bressanelli, S. Polymerases of hepatitis C viruses and flaviviruses: Structural and mechanistic insights and drug development. Antivir. Res. 2014, 105, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Groarke, J.M.; Young, D.C.; Kuhn, R.J.; Smith, J.L.; Pevear, D.C.; Rossmann, M.G. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc. Natl. Acad. Sci. USA 2004, 101, 4425–4430. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.J.; Waksman, G. Structure and mechanism of DNA polymerases. Adv. Protein Chem. 2005, 71, 401–440. [Google Scholar] [PubMed]
- Doublie, S.; Sawaya, M.R.; Ellenberger, T. An open and closed case for all polymerases. Structure 1999, 7, R31–R35. [Google Scholar] [CrossRef]
- Wendt, A.; Adhoute, X.; Castellani, P.; Oules, V.; Ansaldi, C.; Benali, S.; Bourliere, M. Chronic hepatitis c: Future treatment. Clin. Pharmacol. 2014, 6, 1–17. [Google Scholar] [PubMed]
- Larrey, D.; Lohse, A.W.; de Ledinghen, V.; Trepo, C.; Gerlach, T.; Zarski, J.P.; Tran, A.; Mathurin, P.; Thimme, R.; Arasteh, K.; et al. Rapid and strong antiviral activity of the non-nucleosidic NS5B polymerase inhibitor BI 207127 in combination with peginterferon α 2a and ribavirin. J. Hepatol. 2012, 57, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, I.; Pockros, P.J.; Lalezari, J.; Lawitz, E.; Rodriguez-Torres, M.; DeJesus, E.; Haas, F.; Martorell, C.; Pruitt, R.; Purohit, V.; et al. Virologic response rates following 4 weeks of filibuvir in combination with pegylated interferon α-2a and ribavirin in chronically-infected HCV genotype-1 patients. J. Hepatol. 2010, 52, S465–S465. [Google Scholar]
- Rodriguez-Torres, M.; Lawitz, E.; Conway, B.; Kaita, K.; Sheikh, A.M.; Ghalib, R.; Adrover, R.; Cooper, C.; Silva, M.; Rosario, M.; et al. Safety antiviral activity of the HCV non-nucleoside polymerase inhibitor VX-222 in treatment-naive genotype 1 HCV-infected patients. J. Hepatol. 2010, 52, S14–S14. [Google Scholar] [CrossRef]
- Lawitz, E.; Rodriguez-Torres, M.; Rustgi, V.K. Safety and antiviral activity of ana 598 in combination with pegylated interferon α-2a plus ribavirin in treatment-naive genotype 1 chronic HCV patients. J. Hepatol. 2010, 52, 334A–335A. [Google Scholar] [CrossRef]
- Lawitz, E.; Jacobson, I.; Godofsky, E.; Foster, G.R.; Flisiak, R.; Bennett, M.; Ryan, M.; Hinkle, J.; Simpson, J.; McHutchison, J.; et al. A phase 2b trial comparing 24 to 48 weeks treatment with tegobuvir (GS-9190)/PEG/RBV to 48 weeks treatment with PEG/RBV for chronic genotype 1 HCV infection. J. Hepatol. 2011, 54, S181–S181. [Google Scholar] [CrossRef]
- Gane, E.J.; Stedman, C.A.; Hyland, R.H. Nucleotide polymerase inhibi- tor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 2013, 368, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Jensen, D.; Herring, R., Jr. Efficacy and safety of mericitabine in combination with PEG-IFN α-2a/RBV in G1/4 treatment naive HCV patients: Final analysis from the propel study. J. Hepatol. 2012, 56, S481–S482. [Google Scholar] [CrossRef]
- Brown, J.A.; Thorpe, I.F. Dual allosteric inhibitors jointly modulate protein structure and dynamics in the hepatitis c virus polymerase. Biochemistry 2015, 54, 4131–4141. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, J.A. Inhibitors of hcv ns5b polymerase. Part 1: Evaluation of the southern region of (2Z)-2-(benzoylamino)-3-(5-phenyl-2-furyl)acrylic acid. Bioorg. Med. Chem. Lett. 2005, 15, 2481–2486. [Google Scholar] [CrossRef] [PubMed]
- Wang, M. Non-nucleoside analogue inhibitors bind to an allosteric site on hcv ns5b polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 2003, 278, 9489–9495. [Google Scholar] [CrossRef] [PubMed]
- Ontoria, J.M.; Rydberg, E.H.; Carfi, A. Identification and biological evaluation of a series of 1H-benzo[de]isoquinoline-1,3(2H)-diones as hepatitis C virus NS5B polymerase inhibitors. J. Med. Chem. 2009, 52, 5217–5227. [Google Scholar] [CrossRef] [PubMed]
- Nyanguile, O.; Pauwels, F.; van den Broeck, W.; Boutton, C.W.; Quirynen, L.; Ivens, T.; van der Helm, L.; Vandercruyssen, G.; Mostmans, W.; Delouvroy, F.; et al. 1,5-Benzodiazepines, a novel class of hepatitis C virus polymerase nonnucleoside inhibitors. Antimicrob. Agents Chemother. 2008, 52, 4420–4431. [Google Scholar] [CrossRef] [PubMed]
- Nyanguile, O.; Devogelaere, B.; Fanning, G.C. 1a/1bsubtype profiling of nonnucleoside polymerase inhibitors of hepatitis C virus. J. Virol. 2010, 84, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Tomei, L.; Altamura, S.; Migliaccio, G. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2003, 77, 13225–13231. [Google Scholar] [CrossRef] [PubMed]
- Boyce, S.E.; Tirunagari, N.; Niedziela-Majka, A.; Perry, J.; Wong, M.; Kan, E.; Lagpacan, L.; Barauskas, O.; Hung, M.; Fenaux, M.; et al. Structural and regulatory elements of HCV NS5B polymerase—B-Loop and C-terminal tail—Are required for activity of allosteric thumb site II inhibitors. PLoS ONE 2014, 9, e84808. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.Y.; Cheng, H.; Thompson, I.; Chunduru, S.K.; Herrmann, S. Molecular mechanism of a thumb domain hepatitis C virus nonnucleoside RNA-dependent RNA polymerase inhibitor. Antimicrob. Agents Chemother. 2006, 50, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Thorpe, I.F. Molecular simulations illuminate the role of regulatory components of the RNA polymerase from the hepatitis C virus in influencing protein structure and dynamics. Biochemistry 2013, 52, 4541–4552. [Google Scholar] [CrossRef] [PubMed]
- Ando, I.; Adachi, T.; Ogura, N.; Toyonaga, Y.; Sugimoto, K. Preclinical characterization of JTK-853, a novel nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2012, 56, 4250–4256. [Google Scholar] [CrossRef] [PubMed]
- Caillet-Saguy, C.; Simister, P.C.; Bressanellli, S. An objective asessment of conformational variability in complexes of hepatitis C virus polymerase with non-nucleoside inhibitors. J. Mol. Biol. 2011, 414, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, P. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin. Ther. Pat. 2009, 49, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza a polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crepin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sesmero, E.; Thorpe, I.F. Using the Hepatitis C Virus RNA-Dependent RNA Polymerase as a Model to Understand Viral Polymerase Structure, Function and Dynamics. Viruses 2015, 7, 3974-3994. https://doi.org/10.3390/v7072808
Sesmero E, Thorpe IF. Using the Hepatitis C Virus RNA-Dependent RNA Polymerase as a Model to Understand Viral Polymerase Structure, Function and Dynamics. Viruses. 2015; 7(7):3974-3994. https://doi.org/10.3390/v7072808
Chicago/Turabian StyleSesmero, Ester, and Ian F. Thorpe. 2015. "Using the Hepatitis C Virus RNA-Dependent RNA Polymerase as a Model to Understand Viral Polymerase Structure, Function and Dynamics" Viruses 7, no. 7: 3974-3994. https://doi.org/10.3390/v7072808
APA StyleSesmero, E., & Thorpe, I. F. (2015). Using the Hepatitis C Virus RNA-Dependent RNA Polymerase as a Model to Understand Viral Polymerase Structure, Function and Dynamics. Viruses, 7(7), 3974-3994. https://doi.org/10.3390/v7072808