1. Introduction
Humans are the only known natural host of hepatitis B virus (HBV) and so far, the only cell type consistently shown to support productive HBV infection cycle has been hepatocytes [
1]. Such highly restricted host and tissue tropism of HBV makes it ideal as a vector for liver-targeting gene delivery
in vivo. Other viral vectors, including adenovirus and adeno-associated virus, lack such tissue specificity, and infection of human cell types might cause unwanted effects that are difficult to test
in vitro or in animal models [
2]. For
in vitro applications, recombinant HBV vectors expressing easily detectable and quantifiable reporters are powerful tools for addressing important issues such as HBV infection and replication mechanisms.
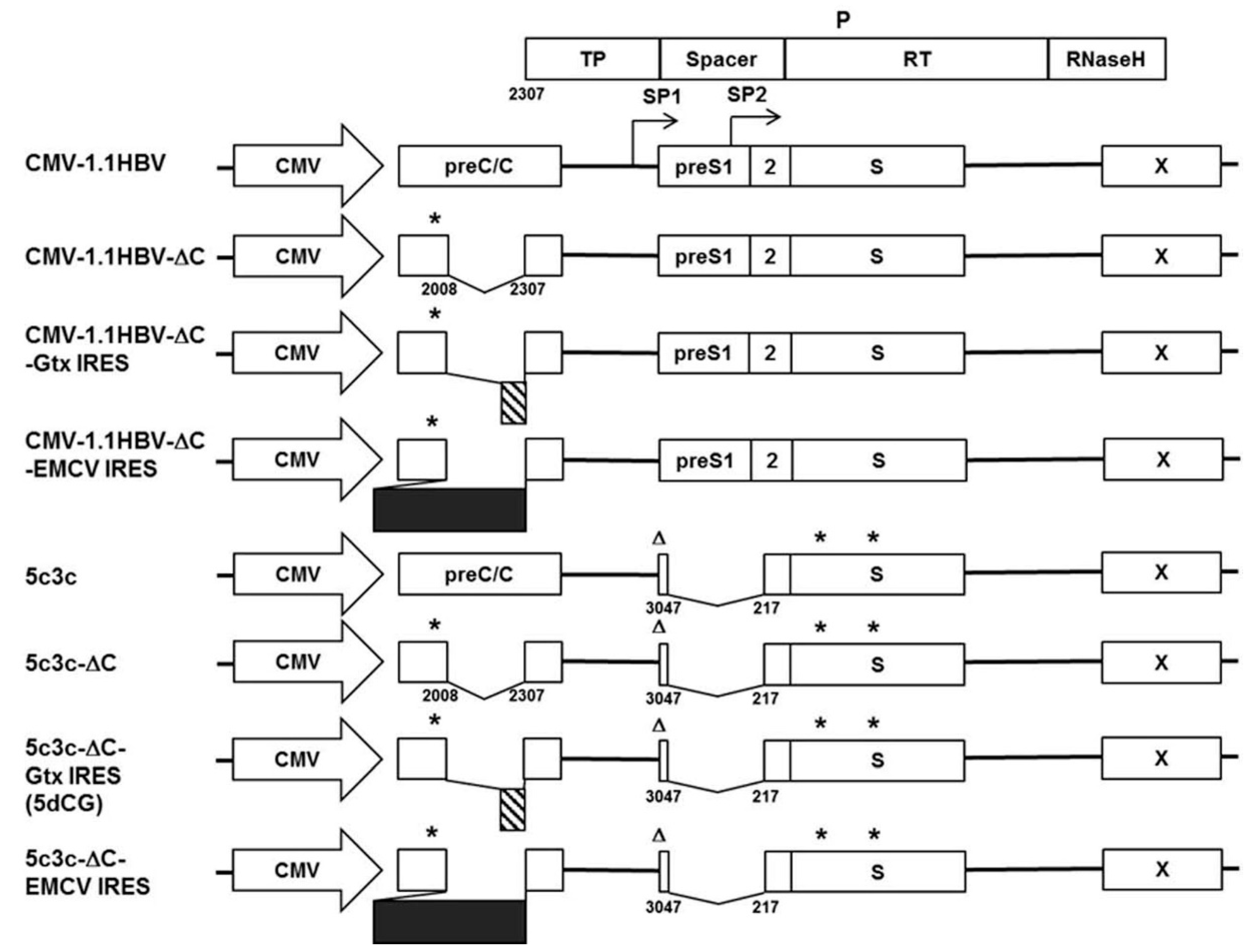
The HBV genome is highly compact and extremely economical in terms of space usage. Four overlapping ORFs (preC/C, P, preS1/preS2/S, X) (
Figure 1) encompass the entire genome, which also contains multiple
cis-acting elements essential for various steps of the viral life cycle [
1,
3]. Partially double-stranded relaxed circular DNA (rcDNA) genomes in mature HBV virions are converted into covalently closed circular DNA (cccDNA) upon infection, which then serves as intranuclear transcription template for all viral RNA species [
1,
3]. Viral polymerase is translated from pregenomic RNA (pgRNA) and binds the latter
in cis to initiate reverse transcription, which is the first step in genome replication [
1,
3,
4]. Packaging of the pgRNA/polymerase complex by viral core proteins, also translated from pgRNA, is required for subsequent steps of replication and eventual formation of rcDNA. Mature capsids are coated by host-derived membranes containing viral large, middle and small (L/M/S) envelope proteins, encoded by the preS1/preS2/S ORF, and bud into ER lumen to produce progeny virions [
1,
3].
The compact and overlapping genome organization, combined with a fairly strict limit on genome size imposed by capsids and polymerase [
5,
6], makes engineering HBV for vector use complicated [
5,
7,
8]. In addition, although
trans-complemented polymerase does support a certain level of replication, recombinant pgRNA encoding functional polymerase that acts
in cis usually replicates with much better efficiency [
4,
8]. Previously, we circumvented these difficulties by modifying a highly replicative clinical isolate of HBV with a large in-frame deletion in preS/polymerase spacer region into a recombinant HBV vector system designated 5c3c (
Figure 1). 5c3c contains a maximized deletion in polymerase spacer region, where cargo gene is inserted, and replicates more efficiently than wild type HBV [
9]. Cargo sequences must be carefully selected or synonymously mutated to avoid introducing stop codons in the overlapping polymerase ORF. 5c3c-based constructs displayed replication efficiencies comparable to wild type HBV and produced mature virions efficiently when
trans-complemented with envelope proteins in transfected cells. Furthermore, in an
in vitro model of HBV infection using primary tupaia hepatocytes (PTH), these 5c3c-based recombinant virions were demonstrated to be infective and deliver expression of cargo protein or RNA genes upon infection [
9].
Despite such demonstrable usability, the 5c3c vector system has limitations that hinder its wider adoption and application. Firstly, the restriction on insertion sequence properties limits possible cargo gene choices. Even when inserted sequences do not introduce premature stop codons into polymerase ORF, unrelated amino acid sequences are inevitably introduced into the polymerase spacer, which may affect polymerase activity and, consequently, replication efficiency. High replication efficiency, however, is a key feature distinguishing 5c3c from most previous recombinant HBV attempts [
5,
7,
8]. Additionally, although mature virion production by 5c3c vectors requires
trans-complemented envelope proteins, replication of 5c3c-based recombinant genomes is self-sufficient and does not rely on co-existing wild type HBV functions [
9]. This may be considered a safety issue in certain
in vivo applications where persistent activity of recombinant HBV is undesirable or even harmful.
In this work, we addressed the above issues with 5c3c by redesigning the cargo gene insertion strategy and engineered a significantly improved derivative vector designated 5dCG. 5dCG is not affected by the insertion sequence restrictions of 5c3c, while retaining a relatively high replication efficiency. Furthermore, cargo gene expression, genome replication and progeny virus production of 5dCG is dependent on trans-complemented core and envelope proteins from co-infecting wild type HBV. Infectivity and cargo gene expression of 5dCG-based recombinant HBV were demonstrated using PTH and properties of 5c3c and 5dCG are comparatively discussed in the context of potential applications.
2. Materials and Methods
2.1. Plasmids
Construction of the recombinant HBV vector 5c3c has been reported previously [
9]. The recombinant 5c3c genome contains an in-frame deletion (nucleotides 3047-3215/1-217) in the polymerase spacer region (
Figure 1) created by maximizing a naturally occurring deletion in the parental clinical isolate (GenBank accession number FJ518810), where cargo sequences could be inserted. In the overlapping preS1/preS2/S ORF, preS1 start codon was mutated from ATG to ACG, which does not change polymerase amino acid at this position. Additionally, S ORF contains 2 premature stop codons inherited from the parental clinical isolate. The genome was cloned into pHY106 vector [
10] downstream of the cytomegalovirus (CMV) promoter to create 5c3c. Wild type HBV genome was amplified from p1.2-HBV containing 1.2 copy of HBV genome [
11] and similarly cloned into pHY106 to create CMV-1.1HBV. Envelope-expressing helper plasmid pLMS encoding wild type envelope proteins was constructed by deleting sequences upstream of the Sp1 promoter from p1.2-HBV. Core-expressing helper plasmid pC was constructed by amplifying core-encoding sequences from 5c3c and cloning into pCDH-CMV-MCS vector (System Biosciences) downstream of CMV promoter.
To test alternative cargo gene insertion sites, 298 bp (nucleotides 2009–2306) of core coding sequences immediately upstream of polymerase start codon were deleted from 5c3c and CMV-1.1HBV to create 5c3c-ΔC and CMV-1.1HBV-ΔC, respectively, using KOD-Plus-Mutagenesis Kit (TOYOBO, Osaka, Japan). A 21 bp linker (
TAACC
CTGCAGGCTC
GCTAGC) containing a termination codon (underlined) that terminates the preceding preC/C ORF as well as
PstI and
NheI recognition sites (underlined and italicized) was introduced in the process to facilitate subsequent insertion of cargo sequences. The 582 bp encephalomyocarditis virus (EMCV) internal ribosomal entry site (IRES) and an 82 bp artificial IRES based on minimal IRES sequences in cellular Gtx gene mRNA, designated (Gtx133-141)2(SII)1β in reference [
12], were chemically synthesized and inserted into 5c3c-ΔC and CMV-1.1HBV-ΔC between the linker and polymerase start codon. The resultant constructs were designated 5c3c-ΔC-EMCV-IRES, 5c3c-ΔC-Gtx-IRES and CMV-1.1HBV-ΔC-EMCV-IRES, CMV-1.1HBV-ΔC-Gtx-IRES, respectively (
Figure 1). For brevity, ‘IRES’ in construct names is sometimes omitted and the vector shown to possess the best properties, 5c3c-ΔC-Gtx-IRES, was abridged as 5dCG and used for subsequent recombinant virus construction. Nucleotide sequences of 5dCG surrounding the cargo gene insertion site are shown in
Figure S1.
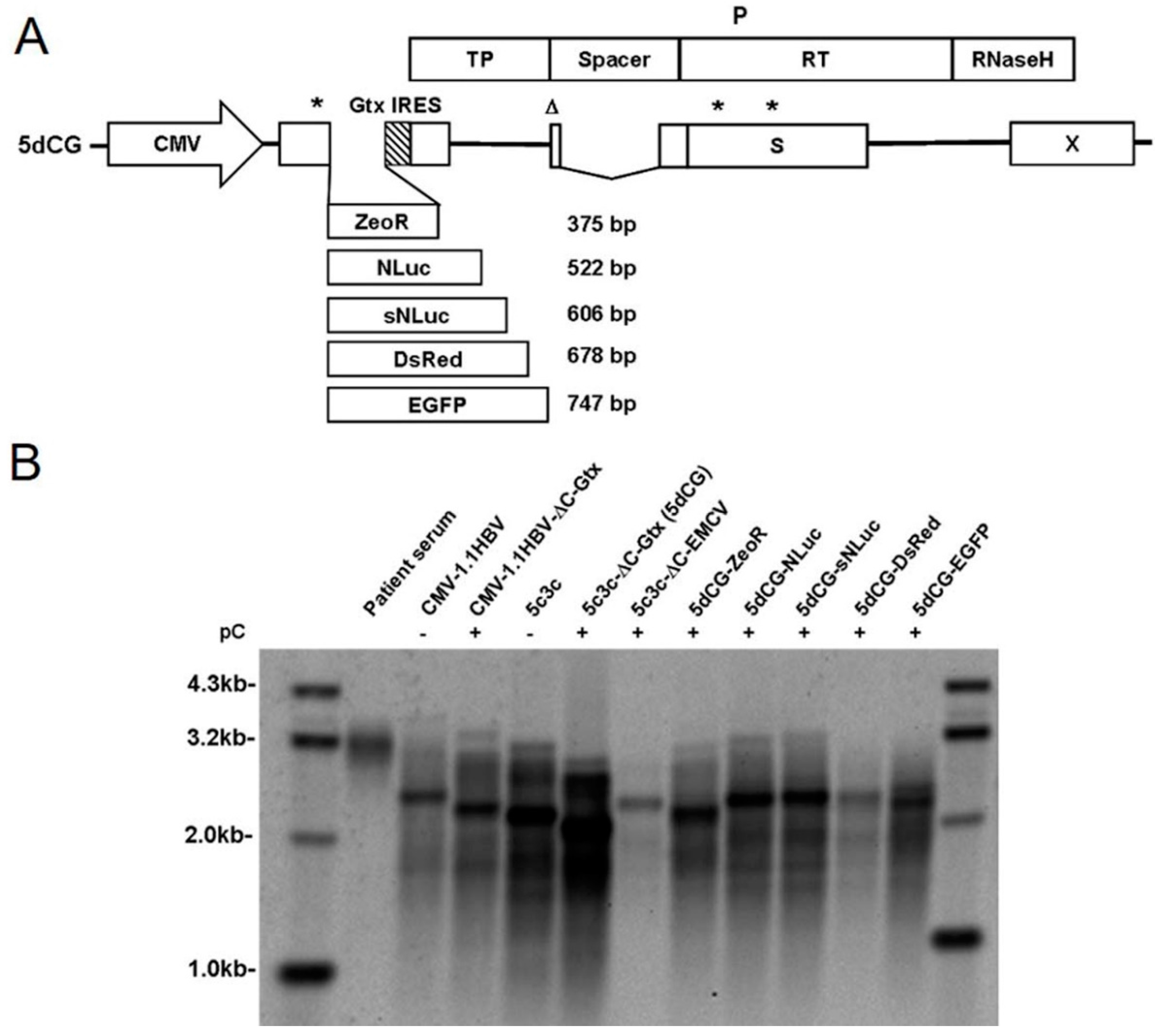
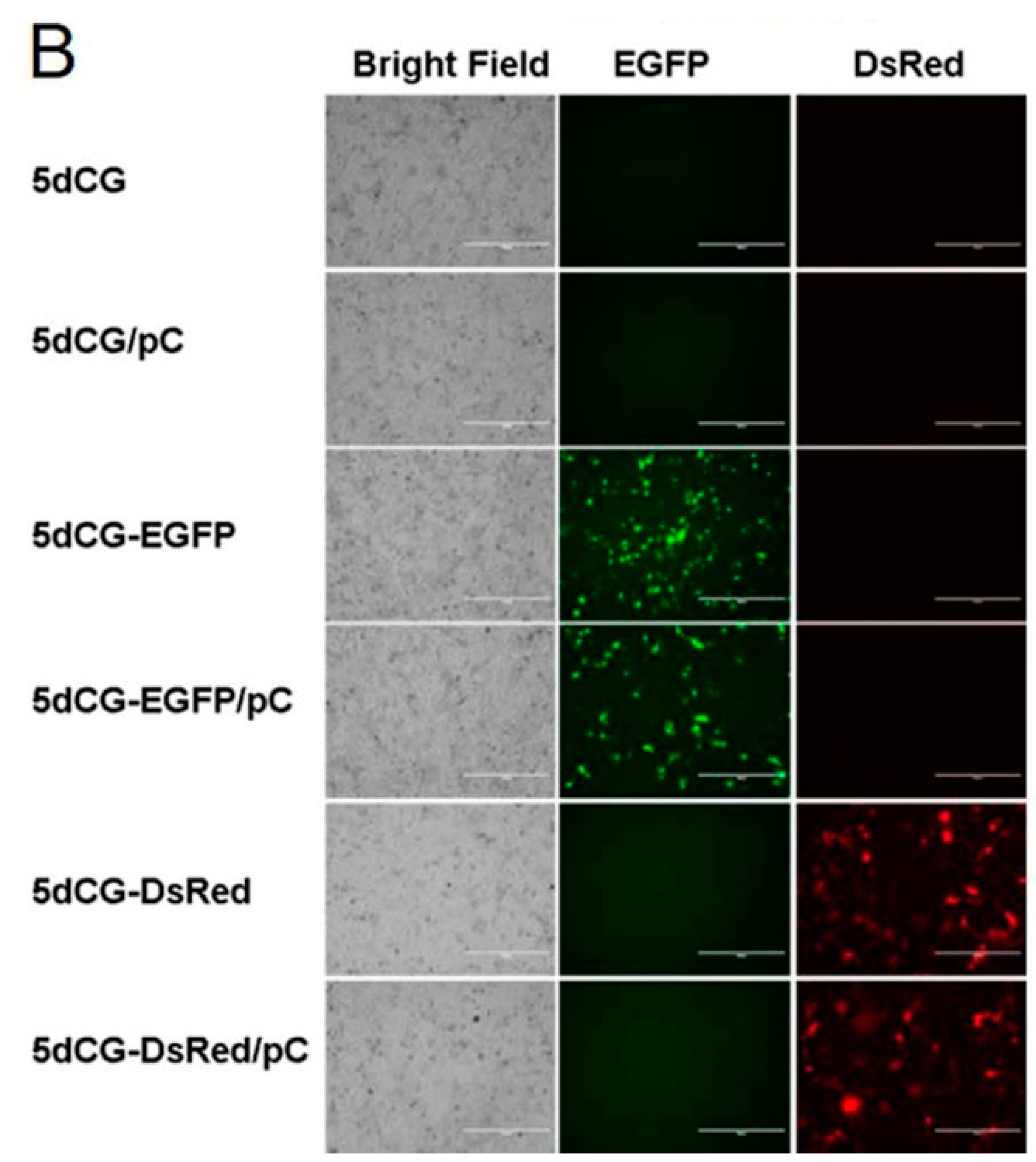
For insertion of protein-encoding genes into 5dCG, sequences encoding zeocin resistance (ZeoR), NanoLuc (NLuc) (Promega, Madison, WI, USA), secreted form of NanoLuc (sNLuc) (Promega), DsRed and EGFP proteins were amplified by PCR from corresponding expression plasmids and inserted into 5dCG using the linker upstream artificial IRES.
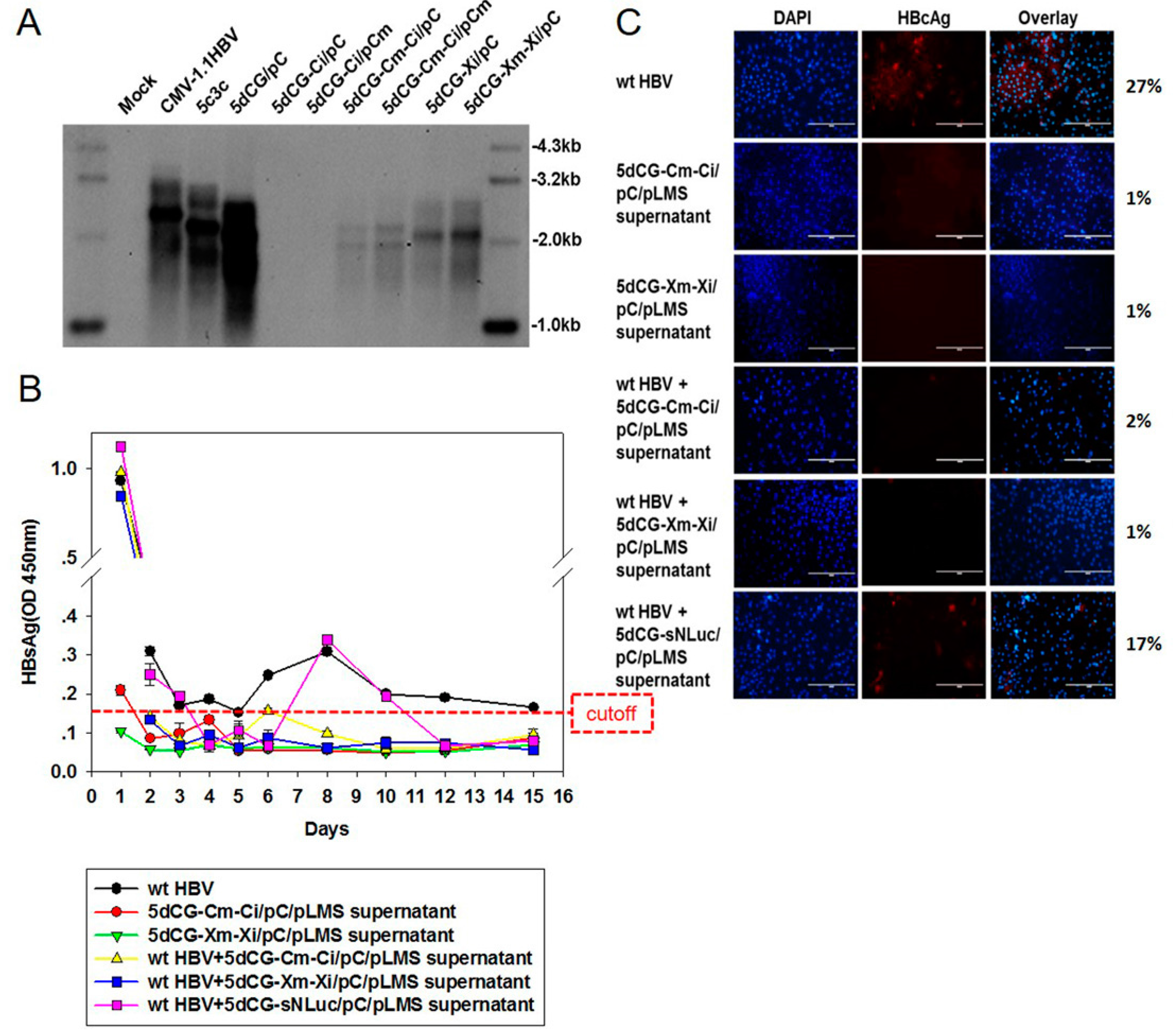
For insertion of short hairpin RNA expression cassettes into 5dCG, sequences containing H1 promoter and downstream shRNA-encoding sequences were amplified from previously reported and verified pSuper based plasmids [
9]. Briefly, Xi targets 5′-CCA
G(A)GT
C(G)TT
G(A)CCCAAGGTCTTACAT-3′ sequences in X ORF, while Ci targets 5′-GATCT
C(T)AA
T(C)CT
C(T)GG
G(A)AA
T(C)CTCA-3′ sequences in C ORF. The underlined nucleotides were mutated to parenthesized nucleotides to create Xi-resistant Xm and Ci-resistant Cm sequences, respectively.
2.2. Cell Culture, Transfection, and HBV Nucleic Acid Analysis
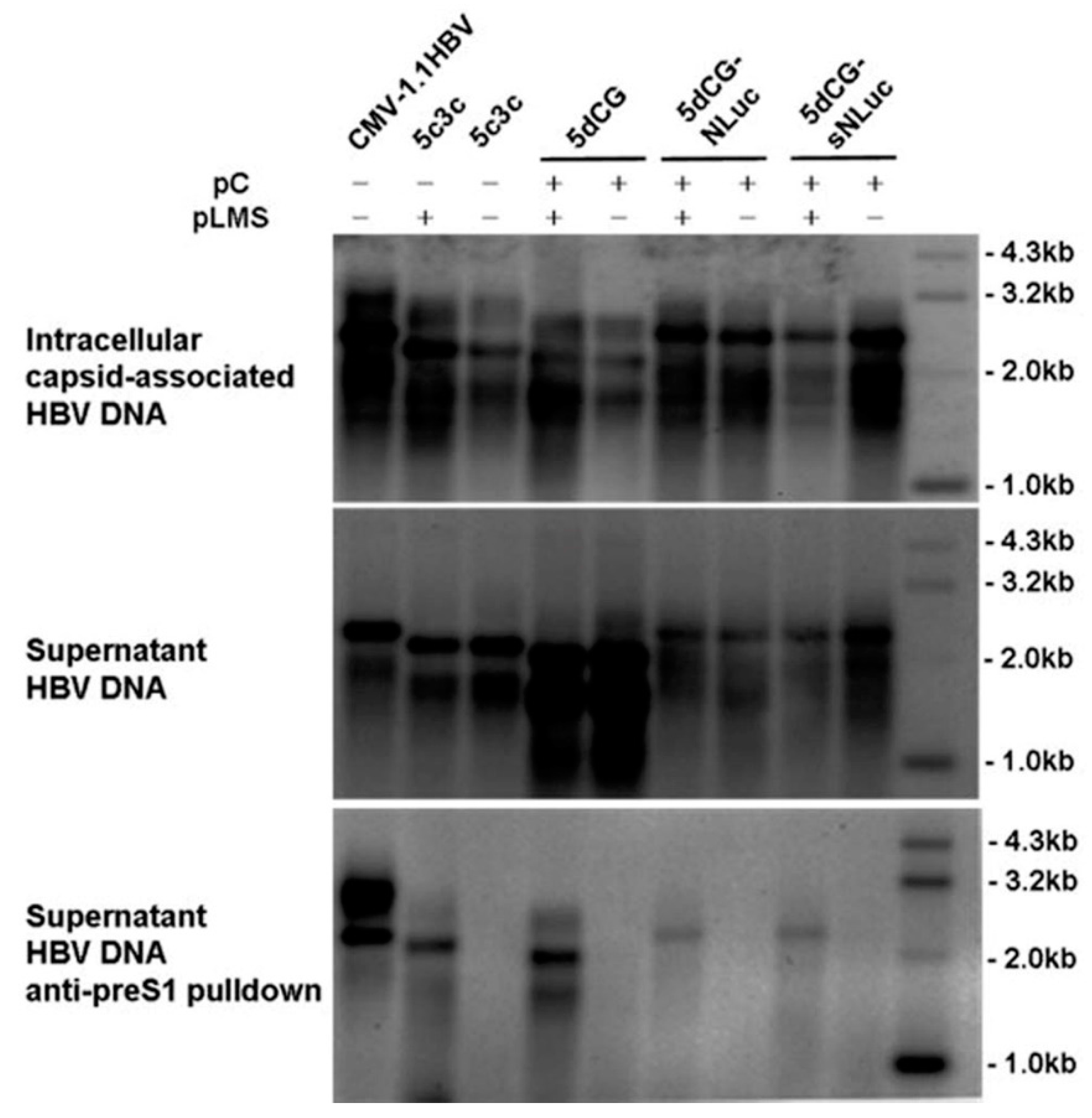
Huh-7 cells were cultured at 37 °C and 5% CO2 in Dulbecco’s modified Eagle medium (DMEM) containing 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin supplemented with 10% fetal bovine serum (all reagents were from Invitrogen, Carlsbad, CA, USA). Transfections were performed at 80 to 90% confluence using plasmid DNA and polyethylenimine (Sigma, St. Louis, MO, USA) at 1:2 ratio. Empty vectors were included whenever necessary to ensure equal amounts of DNA were used in parallel transfections, while control plasmid encoding firefly luciferase reporter (Promega) was co-transfected for transfection efficiency normalization when appropriate. Extraction of intracellular capsid-associated DNA and extracellular capsid- and virion-associated DNA was performed as previously described [
9]. Extracellular Dane particles were enriched by immunoprecipitation using anti-preS1 monoclonal antibody 125E11 (Alpha Inc., Shanghai, China) as previously described [
9] and viral DNA was extracted similarly as above. To demonstrate lack of rcDNA formation by 5c3c-ΔC in the absence of
trans-complemented core, total cellular DNA was extracted using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) and digested with
DpnI (New England BioLabs, Ipswich, MA, USA) to fragment transfected plasmid DNA. Extracted HBV DNA was analyzed via Southern blot using a digoxigenin labeled HBV-specific probe encompassing nucleotides 96 to 1509 of the wild-type HBV genome prepared by using PCR DIG Probe Synthesis Kit (Roche, Mannheim, Germany). Chemiluminescent detection of hybridized probes was performed using DIG High Prime DNA Labeling and Detection Starter Kit II (Roche).
2.3. CsCl Density Gradient Centrifugation
Transfection supernatants, or HBV-infected patient serum, were concentrated using Amicon Ultra-15 (Millipore, Boston, MA, USA) and subjected to CsCl density gradient (1.1 to 1.4 g/cm3) centrifugation at 38,000 rpm for 20 h using SW41 rotor on Beckman L80 (Beckman Coulter, Brea, CA, USA). Fractions were assayed for HBsAg using Abbott Architect system, and HBV DNA was extracted and analyzed using Southern blot as described above.
2.4. Infection of Primary Tupaia Hepatocytes
PTH were prepared following a two-step perfusion protocol as previously described [
13]. Freshly prepared PTH were seeded into collagen-coated 48-well plates (BD Biosciences, Bedford, MA, USA) at 1 × 10
5 cells per well and maintained in Hepato-Stim Hepatocyte Defined medium (BD Biosciences). For infection assays, Amicon Ultra-15-concentrated transfection supernatants containing about 1 × 10
7 HBV genome equivalents (geq), quantitated using a real-time PCR HBV DNA detection kit (Qiagen), were added to each well at 16 h post seeding. After incubation at 37 °C overnight, cells were washed 5 times with PBS and cultured at 37 °C with media changed every day or every other day. For coinfection assays, wild-type HBV virions prepared from patient sera and recombinant HBV particles prepared from transfection supernatants were used at a 1:3 ratio with 1 × 10
7 geq wild type HBV and 3 × 10
7 geq recombinant HBV per well.
2.5. NanoLuc Activity Assay
Culture supernatants of transfected Huh-7 cells or infected PTH were assayed for levels of secreted NanoLuc using Nano-Glo Luciferase Assay System (Promega) following manufacturer’s instructions. For analysis of intracellular NanoLuc activity, cells were washed and diluted in PBS, and then assayed similarly as culture supernatants.
2.6. Immunofluorescence
Fifteen days post infection, PTH were fixed and analyzed using anti-HBcAg (1:700; Dako, Carpinteria, CA, USA) as described previously [
9]. Images were captured using AMG EVOS Fluorescence Microscope (AMG, Mill Creek, WA, USA).
4. Discussion
Although so far limited to laboratory experiments, recombinant HBV has potentially widespread applications both as investigative tools
in vitro and as therapeutic measures
in vivo, owing to the extremely restricted species and tissue tropism of HBV infection and replication. In this work, we modified the cargo gene insertion strategy of our previously established highly replicative 5c3c recombinant HBV vector, in order to solve issues with 5c3c that affect its usability [
9]. By moving the insertion site from preS/polymerase spacer region to core-coding region (
Figure 1), we abolished the restriction imposed on cargo sequences in 5c3c that absolutely dictates the absence of terminator codon in the overlapping polymerase reading frame. Furthermore, introduction of a short artificial IRES upstream of polymerase start codon isolated upstream cargo sequences from downstream polymerase translation and improved replication competence (
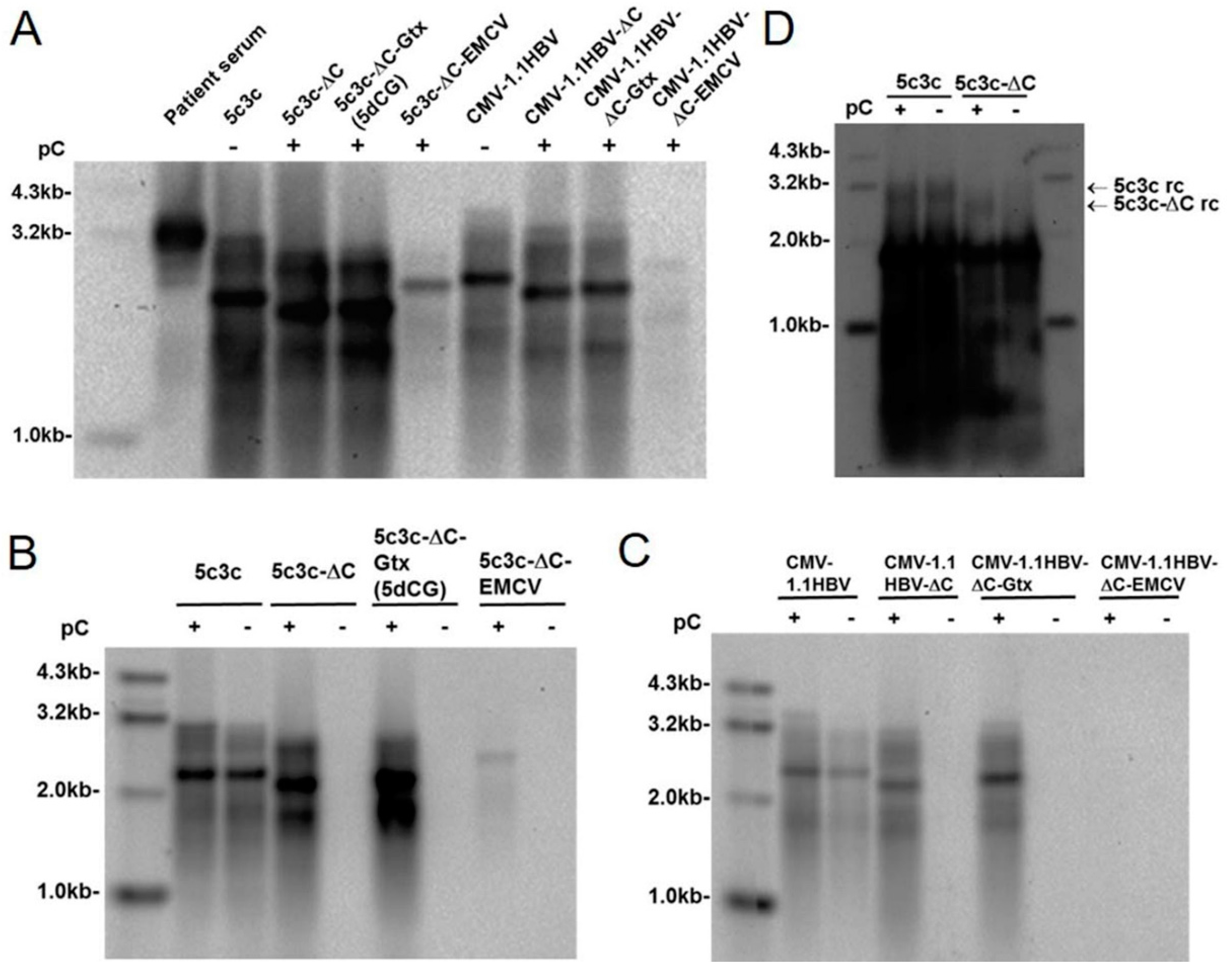
Figure 2B).
Replication efficiency was an issue with a majority of previously reported recombinant HBV designs [
5,
7,
8,
20]. Our previously described vector 5c3c was based on a highly replicative clinical isolate 6898, which harbors an in-frame deletion in preS/polymerase spacer region [
9]. In transfection assays, both 6898 and 5c3c displayed replication efficiencies better than wild type HBV (
Figure 3B) [
9]. Presumably, shortened polymerase spacer plus decreased genome size conferred the replication advantage of 6898 and 5c3c. For this reason, the new vector 5dCG retained the maximized in-frame deletion in preS/polymerase spacer region in 5c3c and replication assay in transfected cells indeed showed that 5dCG vectors replicated much more efficiently than similar vectors derived from wild type HBV with full-length polymerase (
Figure 2A). Retaining this deletion also has the added benefit of freeing up genome spaces for cargo gene insertions. These results reiterated the superiority of 5c3c and 5dCG vectors over previously reported recombinant HBV designs with regard to replication efficiency.
At least two categories of liver-targeting
in vivo applications could be envisioned for recombinant HBV viruses. First, recombinant HBV expressing functional protein or RNA could be used to rectify a potentially life-long pathological condition, e.g., insulin for type I diabetes or coagulation factors for hemophilia [
2]. In such cases, a self-replicating vector like 5c3c that, theoretically, replicates and maintains itself in infected hepatocytes for prolonged periods might be desirable. Another line of applications would involve using recombinant HBV expressing siRNA or miRNA precursors targeting wild type HBV, or immunomodulatory cytokines such as interferon α, as an alternative or supplementary treatment for chronic hepatitis B. In such cases, however, long-lasting persistence and activity of recombinant HBV after wild type HBV infection has been controlled would be undesirable, and may be justifiably considered a safety issue.
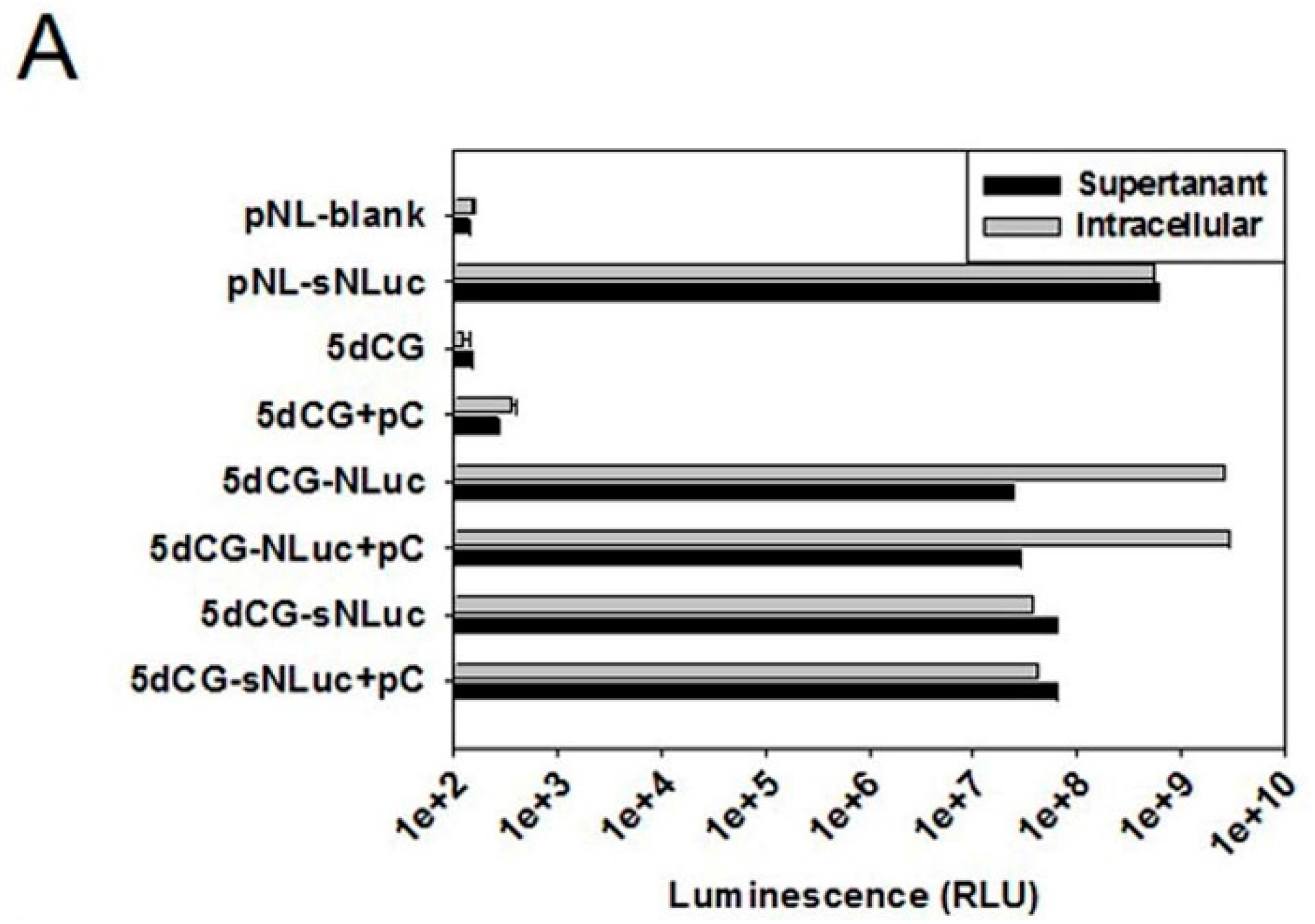
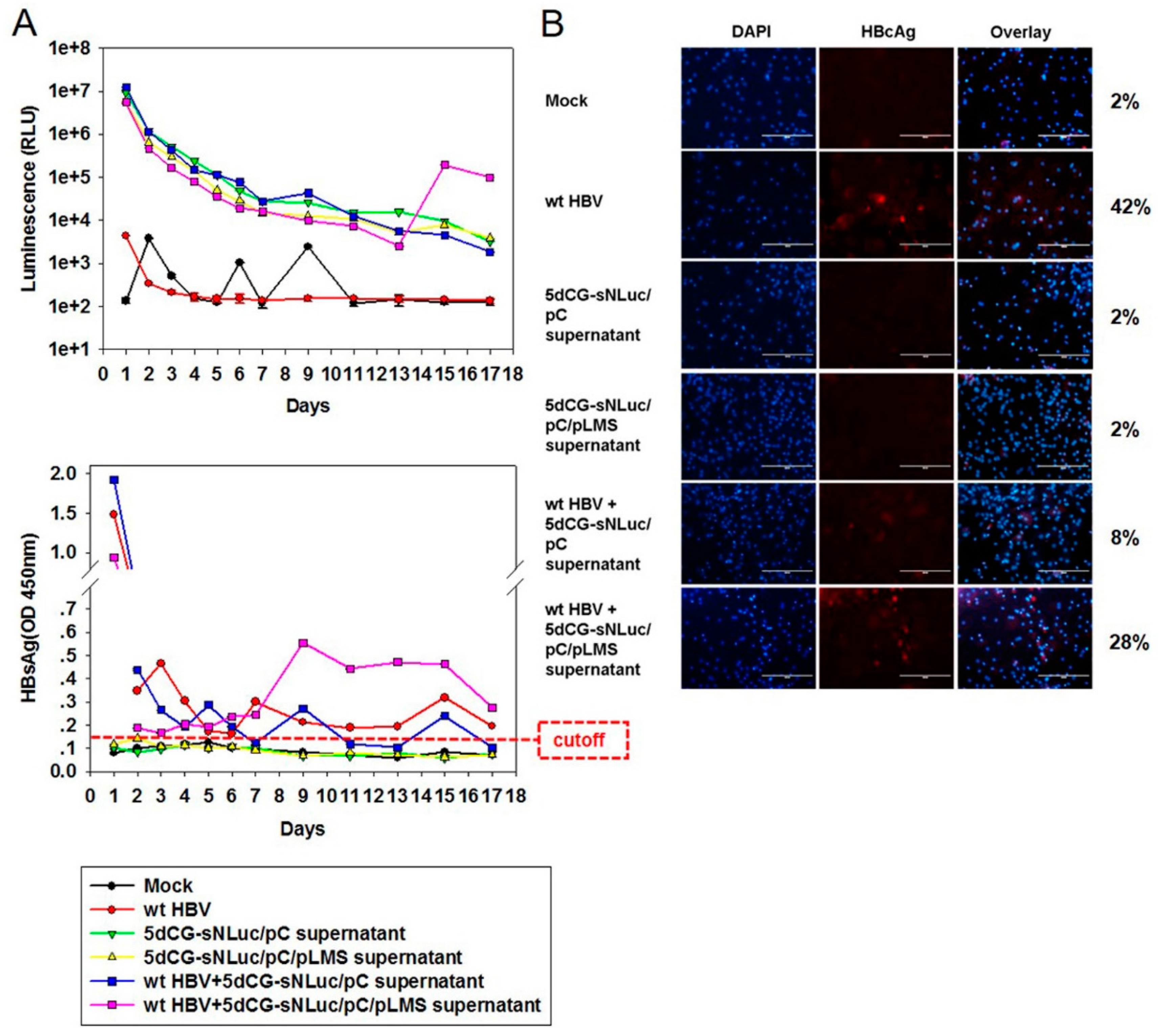
5dCG-based recombinant HBV does not encode functional core or envelope proteins, and would be entirely dependent on
trans-complementation of these viral proteins from co-infecting or super-infecting wild type HBV
in vivo for genome replication and virion production (
Figure 2). Cargo gene expression apparently is also largely dependent on such
trans-complementation (
Figure 6A). In other words, HBV-targeting recombinant viruses, such as 5dCG-Cm-Ci and 5dCG-Xm-Xi, would replicate, expand and effectively produce HBV-targeting cargo gene products, as well as progeny viruses, when and only when there is significant co-existing wild type HBV activity in the same cell. In the absence of such
trans-complementation, lack of self-sufficient replication would render these vectors generally inactive in infected hepatocytes (
Figure 6A). Such “activated-by-target” qualities of 5dCG make it an ideal candidate for HBV-targeting therapeutic applications
in vivo.
Before any human application could be attempted, recombinant HBV would need to be thoroughly studied for activity and long-term persistence in animal models. Unfortunately for HBV, up to now, this would have to rely on either chimpanzees or immuno-deficient mice harboring human hepatocytes, both of which are too expensive to allow studies involving large numbers of animals. Therefore, reporter viruses enabling non-invasive detection of recombinant HBV infection and expression would be invaluable in such cases. For replicating vector 5c3c, we reported the construction of 5c3c-DsRed virus [
9], which could potentially allow detection of infected hepatocytes using
in vivo fluorescence imaging. Similarly, for the non-self-replicating vector 5dCG, we constructed recombinant viruses expressing fluorescent proteins DsRed and EGFP. We also constructed 5dCG-sNL expressing a secreted form of NanoLuc luciferase, which would enable detecting recombinant HBV infection and activity by directly analyzing serum of infected animals. Furthermore, since 5dCG imposes no particular restriction on cargo gene sequences, other reporters allowing more convenient and more sensitive detection and measurement could be easily tested using 5dCG.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







