
Replication-Competent Influenza A Viruses Expressing Reporter Genes



Abstract
:
1. Introduction
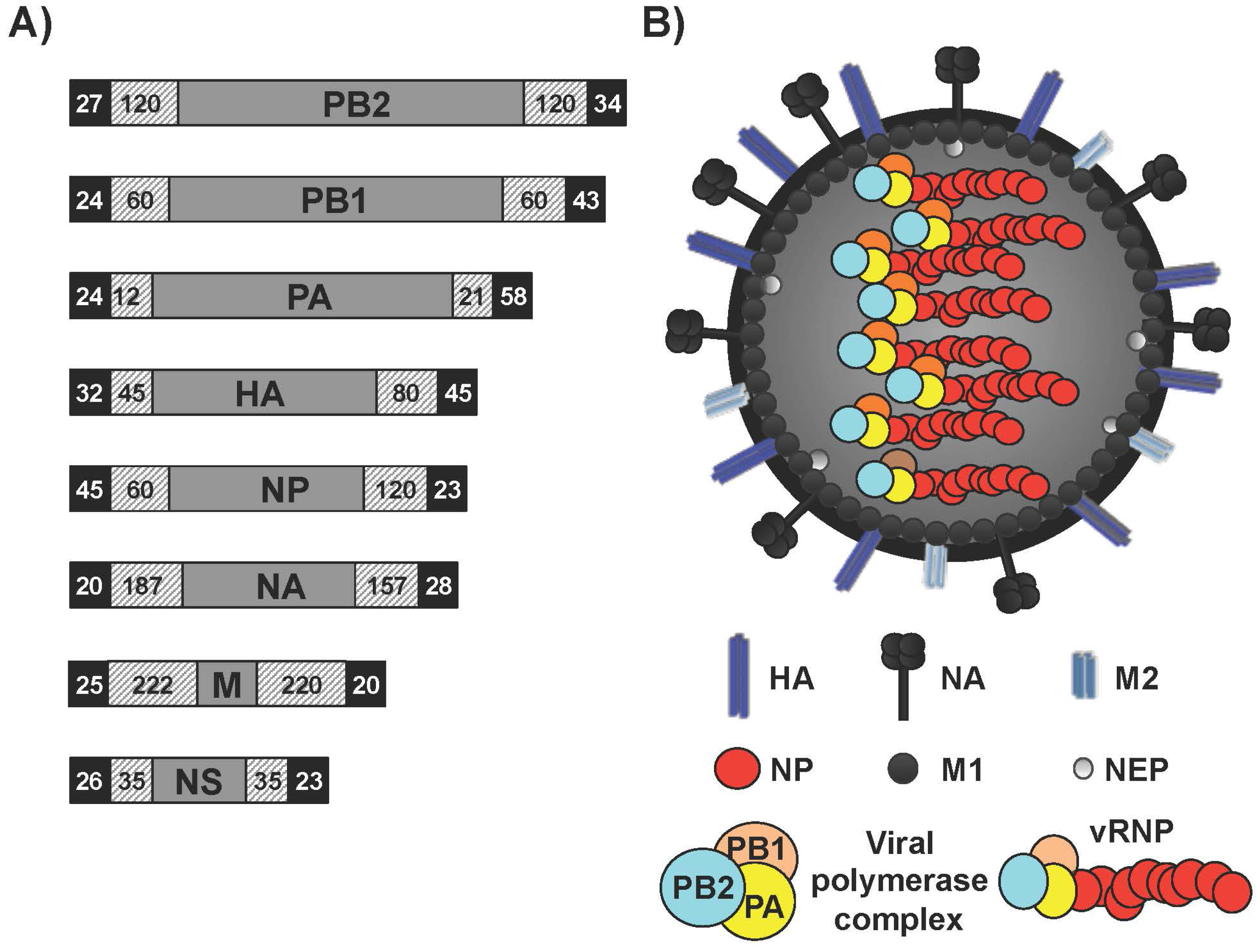
1.1. Influenza A Virus
1.2. Comparison of Fluorescent and Luciferase Reporter Genes
2. Generation of Replication-Competent IAV Harboring Reporter Genes
2.1. Reporter-Expressing IAV Containing the Foreign Gene in the PB2 Segment
2.2. IAV Containing a Viral Polymerase Subunit Fused to the Reporter Gene
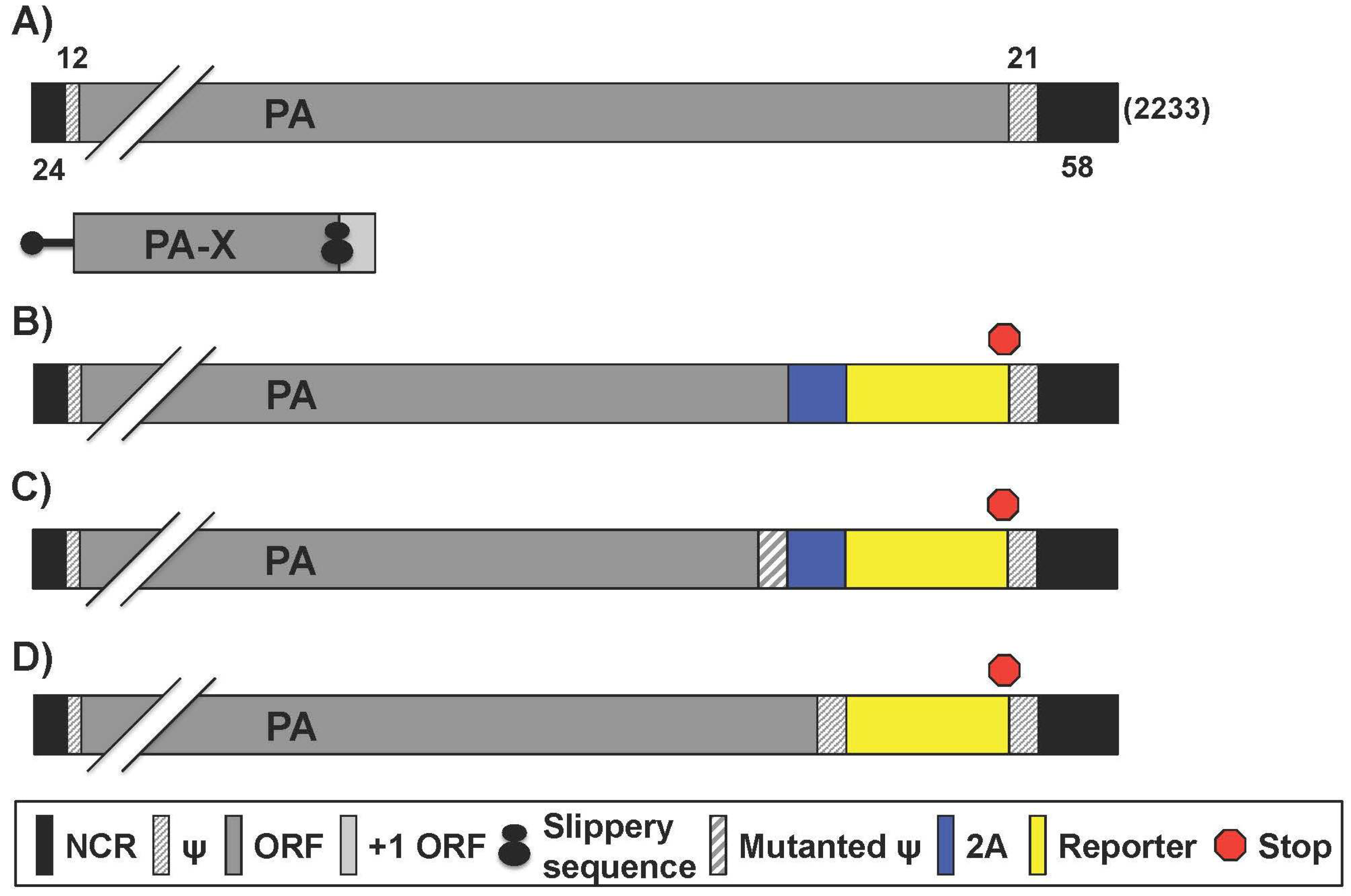
2.3. Reporter IAV Containing a Recombinant PA Segment Harboring the Foreign Gene
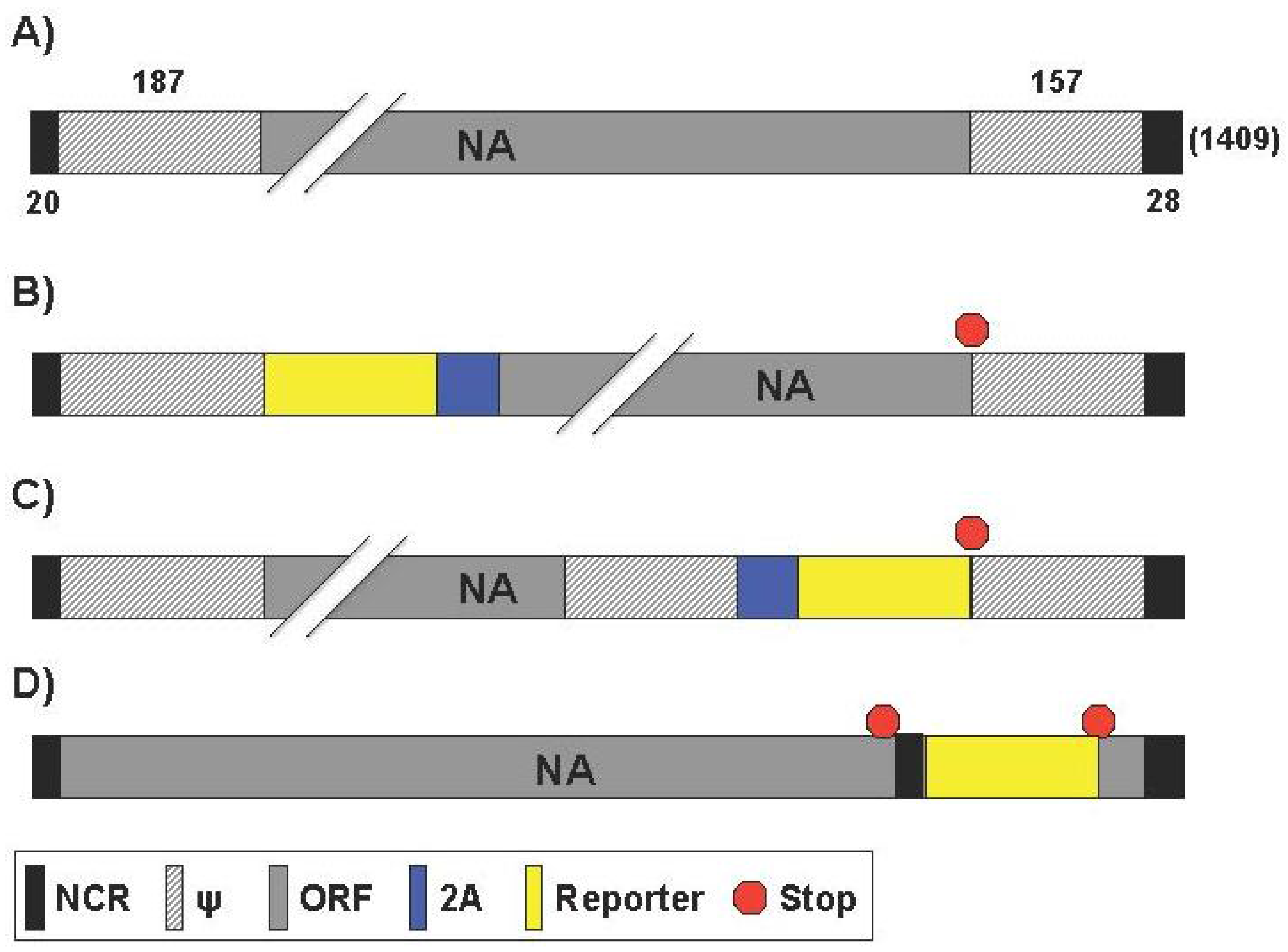
2.4. Generation of Reporter-Expressing IAV Containing a Modified NA Segment
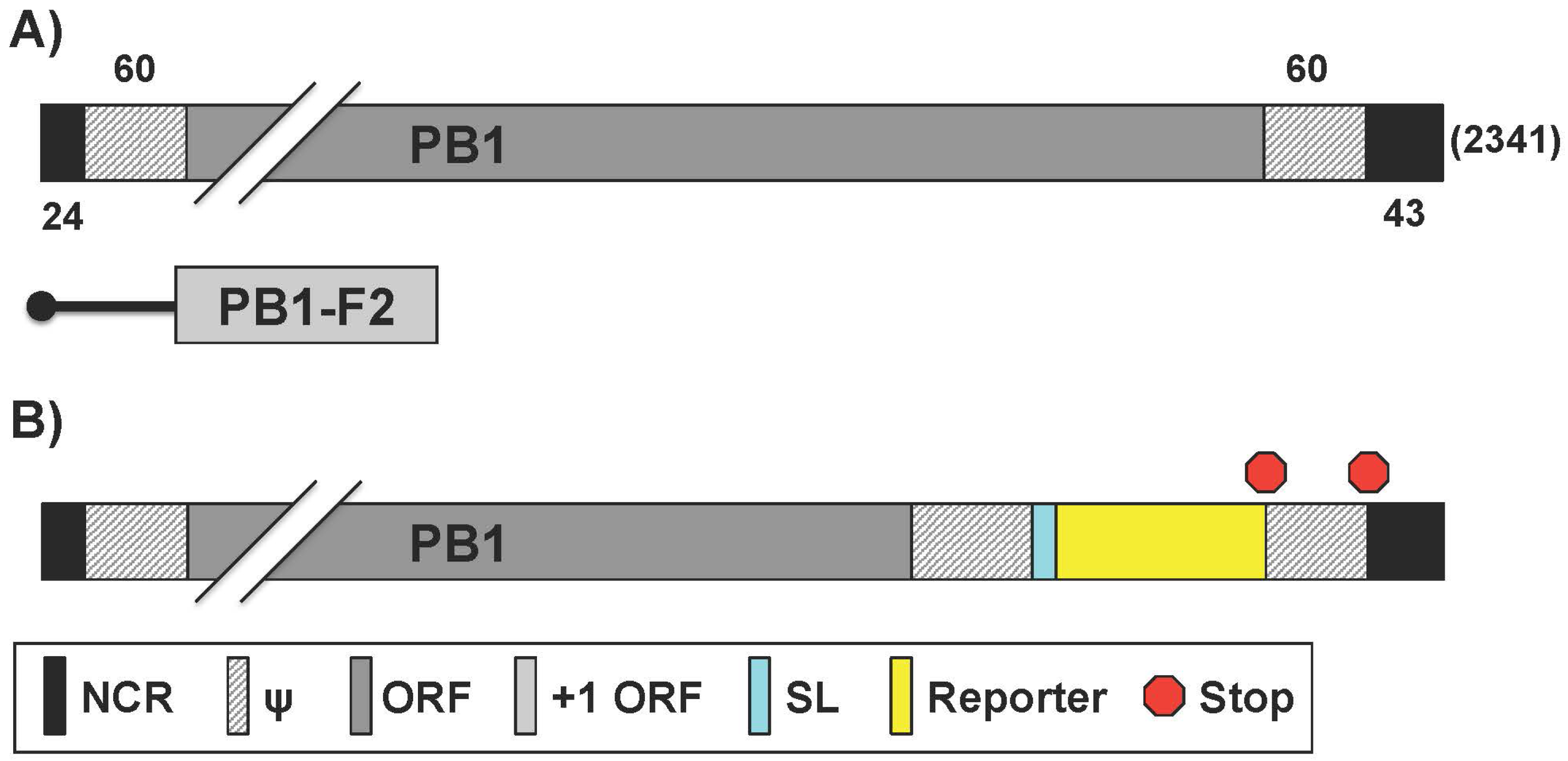
2.5. Generation of Reporter-Expressing IAV by Rearrangement of the PB1 and NS Viral Segment
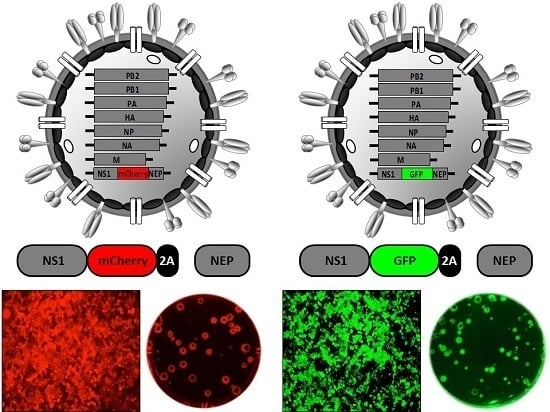
2.6. Generation of Reporter-Expressing, Replicating-Competent IAV Containing a Recombinant NS Segment Harboring the Foreign Gene
3. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| WSN | A/WSN/1933 H1N1 |
| PR8 | A/Puerto Rico/8/1934 |
| pH1N1 | A/California/04/2009 H1N1 |
| Neth602 | A/Netherlands/602/2009 H1N1 |
| Ind5 | A/Indonesia/5/2005 H5N1 |
| Anh1 | A/Anhui/1/2013 H7N9 |
| HK99 | A/Guinea Fowl/Hong Kong/WF10/1999 H9N2 |
| VN1203 | A/Vietnam/1203/2004 H5N1 |
| maxGFP | advanced version of eGFP |
| Venus | advanced version of yellow fluorescent protein |
| aa | amino acid |
| 2A | autocleavage 2A site |
| BMDCs | bone marrow-derived DCs |
| CRS | caspase recognition site |
| CAT | chloramphenicol acetyl transferase |
| DCs | dendritic cells |
| dPR | duplicated packaging region |
| EM | electron microscopy |
| ER | endoplasmic reticulum |
| eCFP | enhanced cyan fluorescent protein |
| eGFP | enhanced green fluorescent protein |
| fRFP | far-red fluorescent protein |
| FFluc | firefly luciferase |
| FACS | fluorescence-activated cell sorting |
| FMDV | foot-and-mouth disease virus |
| Gluc | Gaussia luciferase |
| GFP | green fluorescent protein |
| HA | hemagglutinin |
| HTS | high-throughput screening |
| IAV | influenza A virus |
| BM2 | influenza B virus M2 protein |
| IL-2 | interleukin-2 |
| IVIS | in vivo imaging system |
| MDKC | Madin-Darby canine kidney epithelial cells |
| M1 | matrix 1 protein |
| M2 | matrix 2 protein |
| M2e | matrix 2 protein ectodomain |
| MFI | mean fluorescent intensity |
| Timer | modified DsRed fluorescent protein Timer |
| MAbs | monoclonal antibodies |
| mCherry | monomeric cherry fluorescent protein |
| MLD50 | mouse lethal dose-50 |
| NLuc | NanoLuc |
| iRFP | near-infrared fluorescent protein |
| NA | neuraminidase |
| NAb | neutralizing antibody |
| Ncd protein | nonclaret disjunctional protein |
| NCRs | non-coding regions |
| NS1 | non-structural protein 1 |
| NEP | nuclear export protein |
| NP | nucleoprotein |
| nt | nucleotide |
| ORF | open reading frame |
| ψ | packaging signals |
| PFU | plaque forming units |
| PTV-1 | polymerase acidic (PA) and basic (PB1 and PB2) subunits, porcine teschovirus-1 |
| PET/CT | positron emission tomography-computed tomography |
| PKR | protein kinase R |
| DsRed | red fluorescent protein of Discosoma |
| RIG-I | retinoic acid inducible gene-1 |
| RdRp | RNA-dependent RNA polymerase |
| SL | short linker |
| SAM | splice acceptor mutation |
| TCID50 | tissue culture infectious dose 50 |
| TLR7 | toll-like receptor 7 |
| IFN | type I interferon |
| IFITM proteins | type I IFN-induced transmembrane proteins |
| UTR | untranslated region |
| vRNA | viral RNA |
| vRNP | viral ribonucleoprotein |
| WT | wild-type |
References
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Pritlove, D.C.; Fodor, E.; Seong, B.L.; Brownlee, G.G. In vitro transcription and polymerase binding studies of the termini of influenza A virus cRNA: Evidence for a cRNA panhandle. J. Gen. Virol. 1995, 76, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Flick, R.; Neumann, G.; Hoffmann, E.; Neumeier, E.; Hobom, G. Promoter elements in the influenza vRNAterminal structure. RNA 1996, 2, 1046–1057. [Google Scholar] [PubMed]
- Medina, R.A.; Garcia-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef] [PubMed]
- WHO. fluenza (Seasonal) Fact Sheet No. 211; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K. Influenza hemagglutinin attachment to target cells: ‘Birds do it, we do it...’. Future Virol. 2006, 1, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.N.; Colman, P.M.; van Donkelaar, A.; Blick, T.J.; Sahasrabudhe, A.; McKimm-Breschkin, J.L. Structural evidence for a second sialic acid binding site in avian influenza virus neuraminidases. Proc. Natl. Acad. Sci. USA 1997, 94, 11808–11812. [Google Scholar] [CrossRef] [PubMed]
- Varghese, J.N.; McKimm-Breschkin, J.L.; Caldwell, J.B.; Kortt, A.A.; Colman, P.M. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins 1992, 14, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Cooper, K.L.; Tappenden, P.; Rees, A.; Simpson, E.L.; Read, R.C.; Nicholson, K.G. Oseltamivir, zanamivir and amantadine in the prevention of influenza: A systematic review. J. Infect. 2011, 62, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Oxford, J.S.; Mann, A.; Lambkin, R. A designer drug against influenza: The NA inhibitor oseltamivir (Tamiflu). Expert Rev. Anti Infect. Ther. 2003, 1, 337–342. [Google Scholar] [PubMed]
- Resa-Infante, P.; Jorba, N.; Coloma, R.; Ortin, J. The influenza virus rna synthesis machine: Advances in its structure and function. RNA Biol. 2011, 8, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Hutchinson, E.C.; Jagger, B.W.; Stuart, A.D.; Kang, Z.H.; Robb, N.; Schwartzman, L.M.; Kash, J.C.; Fodor, E.; Firth, A.E.; et al. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 2012, 8, e1002998. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.; Fodor, E. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 2012, 8, e1003019. [Google Scholar] [CrossRef] [PubMed]
- Hai, R.; Schmolke, M.; Varga, Z.T.; Manicassamy, B.; Wang, T.T.; Belser, J.A.; Pearce, M.B.; Garcia-Sastre, A.; Tumpey, T.M.; Palese, P. Pb1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J. Virol. 2010, 84, 4442–4450. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel pb1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional ns1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Lai, C.J. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 1980, 21, 475–485. [Google Scholar] [CrossRef]
- Smith, G.J.D.; Bahl, J.; Vijaykrishna, D.; Zhang, J.X.; Poon, L.L.M.; Chen, H.L.; Webster, R.G.; Peiris, J.S.M.; Guan, Y. Dating the emergence of pandemic influenza viruses. Proc. Natl. Acad. Sci. USA 2009, 106, 11709–11712. [Google Scholar] [CrossRef] [PubMed]
- Jadhao, S.J.; Nguyen, D.C.; Uyeki, T.M.; Shaw, M.; Maines, T.; Rowe, T.; Smith, C.; Huynh, L.P.; Nghiem, H.K.; Nguyen, D.H.; et al. Genetic analysis of avian influenza A viruses isolated from domestic waterfowl in live-bird markets of Hanoi, Vietnam, preceding fatal H5N1 human infections in 2004. Arch. Virol. 2009, 154, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Kilbourne, E.D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Herfst, S.; Schrauwen, E.J.; Linster, M.; Chutinimitkul, S.; de Wit, E.; Munster, V.J.; Sorrell, E.M.; Bestebroer, T.M.; Burke, D.F.; Smith, D.J.; et al. Airborne transmission of influenza A/h5n1 virus between ferrets. Science 2012, 336, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Watanabe, T.; Hatta, M.; Das, S.C.; Ozawa, M.; Shinya, K.; Zhong, G.; Hanson, A.; Katsura, H.; Watanabe, S.; et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 2012, 486, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Uyeki, T.M.; Cox, N.J. Global concerns regarding novel influenza A (H7N9) virus infections. N. Engl. J. Med. 2013, 368, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Aamir, U.B.; Naeem, K.; Ahmed, Z.; Obert, C.A.; Franks, J.; Krauss, S.; Seiler, P.; Webster, R.G. Zoonotic potential of highly pathogenic avian H7N3 influenza viruses from pakistan. Virology 2009, 390, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef]
- Beigel, J.; Bray, M. Current and future antiviral therapy of severe seasonal and avian influenza. Antivir. Res. 2008, 78, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A. Antiviral response in pandemic influenza viruses. Emerg. Infect. Dis. 2006, 12, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Carrat, F.; Flahault, A. Influenza vaccine: The challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef] [PubMed]
- Beyleveld, G.; White, K.M.; Ayllon, J.; Shaw, M.L. New-generation screening assays for the detection of anti-influenza compounds targeting viral and host functions. Antivir. Res. 2013, 100, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ozawa, M.; Iwatsuki-Horimoto, K.; Horimoto, T.; Kawaoka, Y. Incorporation of influenza A virus genome segments does not absolutely require wild-type sequences. J. Gen. Virol. 2009, 90, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Watanabe, S.; Noda, T.; Fujii, Y.; Kawaoka, Y. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 2003, 77, 10575–10583. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; von Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, S.; Katsura, H.; Zhao, D.; Ozawa, M.; Ando, T.; Shoemaker, J.E.; Ishikawa, I.; Yamada, S.; Neumann, G.; Watanabe, S.; et al. Multi-spectral fluorescent reporter influenza viruses (Color-flu) as powerful tools for in vivo studies. Nat. Commun. 2015, 6, 6600. [Google Scholar] [CrossRef] [PubMed]
- Fiege, J.K.; Langlois, R.A. Investigating influenza A virus infection: Tools to track infection and limit tropism. J. Virol. 2015, 89, 6167–6170. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Rodriguez-Sanchez, I.; Monte, K.; Lenschow, D.J.; Perez, D.R.; Martinez-Sobrido, L. Replication-competent fluorescent-expressing influenza b virus. Virus Res. 2015, 213, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Baker, S.F.; Martinez-Sobrido, L. Replication-competent influenza A viruses expressing a red fluorescent protein. Virology 2014, 476, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Patterson, G.H.; Davidson, M.W. Advances in fluorescent protein technology. J. Cell Sci. 2007, 120, 4247–4260. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.K.; Noguchi, T. Cellular bioluminescence imaging. Cold Spring Harb. Protoc. 2012, 2012. [Google Scholar] [CrossRef]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the Luminous Hydromedusan, Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, M.; De, A. Bioluminescence based in vivo screening technologies. Curr. Opin. Pharmacol. 2012, 12, 592–600. [Google Scholar] [CrossRef] [PubMed]
- De Wet, J.R.; Wood, K.V.; DeLuca, M.; Helinski, D.R.; Subramani, S. Firefly luciferase gene: Structure and expression in mammalian cells. Mol. Cell. Biol. 1987, 7, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Eckert, N.; Wrensch, F.; Gartner, S.; Palanisamy, N.; Goedecke, U.; Jager, N.; Pohlmann, S.; Winkler, M. Influenza A virus encoding secreted gaussia luciferase as useful tool to analyze viral replication and its inhibition by antiviral compounds and cellular proteins. PLoS ONE 2014, 9, e97695. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Poole, D.S.; Jeffery, J.J.; Sheahan, T.P.; Creech, D.; Yevtodiyenko, A.; Peat, A.J.; Francis, K.P.; You, S.; Mehle, A. Multi-modal imaging with a toolbox of influenza A reporter viruses. Viruses 2015, 7, 5319–5327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Doyle, T.C.; Coquoz, O.; Kalish, F.; Rice, B.W.; Contag, C.H. Emission spectra of bioluminescent reporters and interaction with mammalian tissue determine the sensitivity of detection in vivo. J. Biomed. Opt. 2005, 10, 41210. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Moser, L.A.; Poole, D.S.; Mehle, A. Highly sensitive real-time in vivo imaging of an influenza reporter virus reveals dynamics of replication and spread. J. Virol. 2013, 87, 13321–13329. [Google Scholar] [CrossRef] [PubMed]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, O.G.; Fodor, E. Functional association between viral and cellular transcription during influenza virus infection. Rev. Med. Virol. 2006, 16, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martinez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Avilov, S.V.; Moisy, D.; Munier, S.; Schraidt, O.; Naffakh, N.; Cusack, S. Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle. J. Virol. 2012, 86, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo-Chaigne, B.; Naffakh, N.; van der Werf, S. Comparative analysis of the ability of the polymerase complexes of influenza viruses type A, B and C to assemble into functional rnps that allow expression and replication of heterotypic model RNA templates in vivo. Virology 1999, 265, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Avilov, S.V.; Moisy, D.; Naffakh, N.; Cusack, S. Influenza A virus progeny vrnp trafficking in live infected cells studied with the virus-encoded fluorescently tagged PB2 protein. Vaccine 2012, 30, 7411–7417. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; Prekeris, R. Polarized endocytic transport: The roles of rab11 and rab11-fips in regulating cell polarity. Histol. Histopathol. 2009, 24, 1171–1180. [Google Scholar] [PubMed]
- Eisfeld, A.J.; Kawakami, E.; Watanabe, T.; Neumann, G.; Kawaoka, Y. Rab11a is essential for transport of the influenza virus genome to the plasma membrane. J. Virol. 2011, 85, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kawaguchi, A.; Takizawa, N.; Morikawa, Y.; Momose, F.; Nagata, K. Involvement of vesicular trafficking system in membrane targeting of the progeny influenza virus genome. Microbes Infect. 2010, 12, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Bruce, E.A.; Digard, P.; Stuart, A.D. The rab11 pathway is required for influenza A virus budding and filament formation. J. Virol. 2010, 84, 5848–5859. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Leyva-Grado, V.H.; Tan, G.S.; Eggink, D.; Hai, R.; Palese, P. in vivo bioluminescent imaging of influenza A virus infection and characterization of novel cross-protective monoclonal antibodies. J. Virol. 2013, 87, 8272–8281. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Weisshaar, M.; Lamb, K.; Chung, H.K.; Lin, M.Z.; Plemper, R.K. Replication-competent influenza virus and respiratory syncytial virus luciferase reporter strains engineered for co-infections identify antiviral compounds in combination screens. Biochemistry 2015, 54, 5589–5604. [Google Scholar] [CrossRef] [PubMed]
- Breen, M.; Nogales, A.; Baker, S.F.; Perez, D.R.; Martinez-Sobrido, L. Replication-competent influenza A and b viruses expressing a fluorescent dynamic timer protein for in vitro and in vivo studies. PLoS ONE 2016, 11, e0147723. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.A.; Meliopoulos, V.A.; Savage, C.; Livingston, B.; Mehle, A.; Schultz-Cherry, S. Visualizing real-time influenza virus infection, transmission and protection in ferrets. Nat. Commun. 2015, 6, 6378. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Feng, L.; Pan, W.; Dong, Z.; Li, C.; Sun, C.; Chen, L. Generation of replication-competent recombinant influenza A viruses carrying a reporter gene harbored in the neuraminidase segment. J. Virol. 2010, 84, 12075–12081. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, B.; Manicassamy, S.; Belicha-Villanueva, A.; Pisanelli, G.; Pulendran, B.; Garcia-Sastre, A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. USA 2010, 107, 11531–11536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Dong, Z.; Li, F.; Meng, W.; Feng, L.; Niu, X.; Li, C.; Luo, Q.; Li, Z.; Sun, C.; et al. Visualizing influenza virus infection in living mice. Nat. Commun. 2013, 4, 2369. [Google Scholar] [CrossRef] [PubMed]
- Spronken, M.I.; Short, K.R.; Herfst, S.; Bestebroer, T.M.; Vaes, V.P.; van der Hoeven, B.; Koster, A.J.; Kremers, G.J.; Scott, D.P.; Gultyaev, A.P.; et al. Optimisations and challenges involved in the creation of various bioluminescent and fluorescent influenza A virus strains for in vitro and in vivo applications. PLoS ONE 2015, 10, e0133888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Yan, F.; Doronina, V.A.; Escuin-Ordinas, H.; Ryan, M.D.; Brown, J.D. 2A peptides provide distinct solutions to driving stop-carry on translational recoding. Nucleic Acids Res. 2012, 40, 3143–3151. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Hwang, W.C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.M.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Mullarkey, C.E.; Miller, M.S. Measuring the neutralization potency of influenza A virus hemagglutinin stalk/stem-binding antibodies in polyclonal preparations by microneutralization assay. Methods 2015, 90, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Hai, R.; Krammer, F.; Tan, G.S.; Pica, N.; Eggink, D.; Maamary, J.; Margine, I.; Albrecht, R.A.; Palese, P. Influenza viruses expressing chimeric hemagglutinins: Globular head and stalk domains derived from different subtypes. J. Virol. 2012, 86, 5774–5781. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Kunz, A.; Simon, V.A.; Palese, P.; Shaw, M.L. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl. Acad. Sci. USA 2011, 108, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Guo, H.; Albrecht, R.A.; Garcia-Sastre, A.; Topham, D.J.; Martinez-Sobrido, L. Protection against lethal influenza with a viral mimic. J. Virol. 2013, 87, 8591–8605. [Google Scholar] [CrossRef] [PubMed]
- Munier, S.; Rolland, T.; Diot, C.; Jacob, Y.; Naffakh, N. Exploration of binary virus-host interactions using an infectious protein complementation assay. Mol. Cell. Proteom. 2013, 12, 2845–2855. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Bailey, K.W.; Wong, M.H.; Barnard, D.L.; Smee, D.F. In vitro and in vivo influenza virus-inhibitory effects of viramidine. Antivir. Res. 2005, 68, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Pekosz, A.; Shuck, K.; Pinto, L.H.; Lamb, R.A. Influenza A virus M2 ion channel activity is essential for efficient replication in tissue culture. J. Virol. 2002, 76, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E.; Smith, M. The pa subunit is required for efficient nuclear accumulation of the PB1 subunit of the influenza A virus RNA polymerase complex. J. Virol. 2004, 78, 9144–9153. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Baker, S.F.; Domm, W.; Martinez-Sobrido, L. Development and applications of single-cycle infectious influenza A virus (sciIAV). Virus Res 2016, 216, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hong, Y.; Parslow, T.G. Cis-acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.; Lowen, A.C.; Mubareka, S.; Palese, P. Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLoS Pathog. 2009, 5, e1000252. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.S.; Wu, Y.; Wawrzusin, P.; Kabat, J.; Broadbent, A.J.; Lamirande, E.W.; Fodor, E.; Altan-Bonnet, N.; Shroff, H.; Subbarao, K. Influenza A virus assembly intermediates fuse in the cytoplasm. PLoS Pathog. 2014, 10, e1003971. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, R.A.; Garcia-Sastre, A.; Kawaoka, Y.; Barclay, W.S.; Bouvier, N.M.; Brown, I.H.; Capua, I.; Chen, H.; Compans, R.W.; Couch, R.B.; et al. Transmission studies resume for avian flu. Science 2013, 339, 520–521. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.T.; Garcia-Sastre, A.; Manicassamy, B. Insertion of a GFP reporter gene in influenza virus. Curr. Protoc. Microbiol. 2013. [Google Scholar] [CrossRef]
- Kittel, C.; Sereinig, S.; Ferko, B.; Stasakova, J.; Romanova, J.; Wolkerstorfer, A.; Katinger, H.; Egorov, A. Rescue of influenza virus expressing gfp from the NS1 reading frame. Virology 2004, 324, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, D.V.; Nordholm, J.; Dou, D.; Wang, H.; Rossman, J.S.; Daniels, R. The influenza virus neuraminidase protein transmembrane and head domains have coevolved. J. Virol. 2015, 89, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Steukers, L.; Forier, K.; Xiong, R.; Braeckmans, K.; Van Reeth, K.; Nauwynck, H. A beneficiary role for neuraminidase in influenza virus penetration through the respiratory mucus. PLoS ONE 2014, 9, e110026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, A.V.; Naffakh, N.; van der Werf, S.; Escriou, N. Expression of a foreign gene by stable recombinant influenza viruses harboring a dicistronic genomic segment with an internal promoter. Virology 2003, 313, 235–249. [Google Scholar] [CrossRef]
- Machado, A.V.; Naffakh, N.; Gerbaud, S.; van der Werf, S.; Escriou, N. Recombinant influenza A viruses harboring optimized dicistronic na segment with an extended native 5’ terminal sequence: Induction of heterospecific B and T cell responses in mice. Virology 2006, 345, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.R.; Garcia-Sastre, A. H5N1, a wealth of knowledge to improve pandemic preparedness. Virus Res. 2013, 178, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.R.; Lim, W.; Seiler, J.P.; Yi, G.; Peiris, M.; Shortridge, K.F.; Webster, R.G. Role of quail in the interspecies transmission of H9 influenza A viruses: Molecular changes on ha that correspond to adaptation from ducks to chickens. J. Virol. 2003, 77, 3148–3156. [Google Scholar] [CrossRef] [PubMed]
- Pena, L.; Sutton, T.; Chockalingam, A.; Kumar, S.; Angel, M.; Shao, H.; Chen, H.; Li, W.; Perez, D.R. Influenza viruses with rearranged genomes as live-attenuated vaccines. J. Virol. 2013, 87, 5118–5127. [Google Scholar] [CrossRef] [PubMed]
- Kimble, J.B.; Sorrell, E.; Shao, H.; Martin, P.L.; Perez, D.R. Compatibility of H9N2 avian influenza surface genes and 2009 pandemic H1N1 internal genes for transmission in the ferret model. Proc. Natl. Acad. Sci. USA 2011, 108, 12084–12088. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.C.; Obadan, A.; Lavigne, J.; Chen, H.; Li, W.; Perez, D.R. Genome rearrangement of influenza virus for anti-viral drug screening. Virus Res 2014, 189, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Kittel, C.; Ferko, B.; Kurz, M.; Voglauer, R.; Sereinig, S.; Romanova, J.; Stiegler, G.; Katinger, H.; Egorov, A. Generation of an influenza A virus vector expressing biologically active human interleukin-2 from the ns gene segment. J. Virol. 2005, 79, 10672–10677. [Google Scholar] [CrossRef] [PubMed]
- Gabor, K.A.; Goody, M.F.; Mowel, W.K.; Breitbach, M.E.; Gratacap, R.L.; Witten, P.E.; Kim, C.H. Influenza A virus infection in zebrafish recapitulates mammalian infection and sensitivity to anti-influenza drug treatment. Dis. Models Mech. 2014, 7, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, I.K.; Pillai, P.S.; Iwasaki, A. Efficient influenza A virus replication in the respiratory tract requires signals from TLR7 and RIG-I. Proc. Natl. Acad. Sci. USA 2013, 110, 13910–13915. [Google Scholar] [CrossRef] [PubMed]
- Resa-Infante, P.; Thieme, R.; Ernst, T.; Arck, P.C.; Ittrich, H.; Reimer, R.; Gabriel, G. Importin-alpha7 is required for enhanced influenza A virus replication in the alveolar epithelium and severe lung damage in mice. J. Virol. 2014, 88, 8166–8179. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Manicassamy, B.; Lamb, R.A. Influenza A virus uses intercellular connections to spread to neighboring cells. J. Virol. 2015, 89, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- De Baets, S.; Verhelst, J.; Van den Hoecke, S.; Smet, A.; Schotsaert, M.; Job, E.R.; Roose, K.; Schepens, B.; Fiers, W.; Saelens, X. A GFP expressing influenza A virus to report in vivo tropism and protection by a matrix protein 2 ectodomain-specific monoclonal antibody. PLoS ONE 2015, 10, e0121491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuther, P.; Gopfert, K.; Dudek, A.H.; Heiner, M.; Herold, S.; Schwemmle, M. Generation of a variety of stable influenza A reporter viruses by genetic engineering of the ns gene segment. Sci. Rep. 2015, 5, 11346. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.M.; Williams, M.A.; Lamb, R.A. Eukaryotic coupled translation of tandem cistrons: Identification of the influenza B virus BM2 polypeptide. EMBO J. 1990, 9, 2639–2647. [Google Scholar] [PubMed]
- Basler, C.F.; Reid, A.H.; Dybing, J.K.; Janczewski, T.A.; Fanning, T.G.; Zheng, H.; Salvatore, M.; Perdue, M.L.; Swayne, D.E.; Garcia-Sastre, A.; et al. Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl. Acad. Sci. USA 2001, 98, 2746–2751. [Google Scholar] [CrossRef] [PubMed]
- Helft, J.; Manicassamy, B.; Guermonprez, P.; Hashimoto, D.; Silvin, A.; Agudo, J.; Brown, B.D.; Schmolke, M.; Miller, J.C.; Leboeuf, M.; et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J. Clin. Investig. 2012, 122, 4037–4047. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.M.; Richardson, G.; Zhou, H.; Manicassamy, B.; Garcia-Sastre, A.; Enelow, R.I.; Braciale, T.J. Influenza-infected neutrophils within the infected lungs act as antigen presenting cells for anti-viral CD8(+) T cells. PLoS ONE 2012, 7, e46581. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Park, S.; Lee, I.; Lee, S.; Shin, S.; Won, Y.; Hwang, M.W.; Bae, J.Y.; Heo, J.; Hyun, H.E.; et al. GFP-expressing influenza A virus for evaluation of the efficacy of antiviral agents. J. Microbiol. 2012, 50, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The ifitm proteins mediate cellular resistance to influenza A H1N1 virus, west nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of ifitm-mediated restriction of filoviruses, sars coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.C.; Farzan, M.; Jung, J.U. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Cenedella, R.J. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids 2009, 44, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Terskikh, A.; Fradkov, A.; Ermakova, G.; Zaraisky, A.; Tan, P.; Kajava, A.V.; Zhao, X.; Lukyanov, S.; Matz, M.; Kim, S.; et al. “Fluorescent timer”: Protein that changes color with time. Science 2000, 290, 1585–1588. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.L.; Webster, R.G. Pandemic influenza As a current threat. Curr. Top. Microbiol. Immunol. 2009, 333, 3–24. [Google Scholar] [PubMed]
- Taubenberger, J.K.; Morens, D.M. 1918 influenza: The mother of all pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Basler, C.F.; Williams, B.R.; Silverman, R.H.; Palese, P.; Garcia-Sastre, A. Functional replacement of the carboxy-terminal two-thirds of the influenza A virus NS1 protein with short heterologous dimerization domains. J. Virol. 2002, 76, 12951–12962. [Google Scholar] [CrossRef] [PubMed]
- Neirynck, S.; Deroo, T.; Saelens, X.; Vanlandschoot, P.; Jou, W.M.; Fiers, W. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat. Med. 1999, 5, 1157–1163. [Google Scholar] [PubMed]
- Martinez-Sobrido, L.; Garcia-Sastre, A. Generation of recombinant influenza virus from plasmid DNA. J. Vis. Exp. 2010, 42, e2057. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Neumann, G.; Kawaoka, Y.; Hobom, G.; Webster, R.G. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA 2000, 97, 6108–6113. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Fluorescence | Bioluminescence |
|---|---|---|
| Enzymatic amplification of signal | NO | YES |
| Substrate required for assay | NO | YES |
| High Reproducibility | YES | YES |
| FACS-compatible | YES | NO |
| In vitro applications | YES | YES |
| Ex vivo applications | YES | YES |
| In vivo applications | NO | YES |
| Flexible readout | YES | NO |
| Analysis of individual cells | YES | NO |
| HTS | YES | YES |
| Analysis of intermolecular interactions | YES | NO |
| Detection | Fluorescence | Luminescence |
| Gene | Virus Backbone (1) | Transgene (2) | Insertion Mechanism (3) | Application | Ref. |
|---|---|---|---|---|---|
| PB2 | WSN | Split GFP | Fusion | Virus Biology | [58,61] |
| PB2 | PR8, WSN | Gluc | 2A site | Neutralizing antibodies Antivirals | [66,67] |
| Gene | Virus Backbone (1) | Transgene (2) | Insertion Mechanism (3) | Application | Ref. |
|---|---|---|---|---|---|
| PB2 PB1 PA | WSN | Split Gluc | Fusion | Virus-host interaction | [80] |
| Gene | Virus Backbone (1) | Transgene (2) | Insertion Mechanism (3) | Application | Ref. |
|---|---|---|---|---|---|
| PA | WSN | Nluc | 2A site | Virus biology and transmission | [50,52] |
| PA | pH1N1 | Nluc | 2A site | Virus biology and transmission | [69] |
| PA | PR8, Neth602, Ind5, Anh1 | eGFP, fRFP, iRFP, Gluc, FFluc | 2A site | Virus biology | [73] |
| PA | WSN | GFP | Fusion | Virus biology | [87] |
| Gene | Virus Backbone (1) | Transgene (2) | Insertion Mechanism (3) | Application | Ref. |
|---|---|---|---|---|---|
| NA | PR8 | eGFP | 2A site | Virus biology | [70] |
| NA | PR8 | Gluc | 2A site | Virus biology | [72] |
| NA | WSN | GFP | Viral promoter | Virus biology | [93,94] |
| Gene | Virus Backbone (1) | Transgene (2) | Insertion Mechanism (3) | Application | Ref. |
|---|---|---|---|---|---|
| NS | HK99 VN1203 | GFP, Gluc | Genome rearrangement | Vaccine | [97] |
| NS | pH1N1 | GFP, Gluc | Genome rearrangement | Antivirals | [99] |
| Gene | Virus Backbone (1) | Transgene (2) | Insertion Mechanism (3) | Application | Ref. |
|---|---|---|---|---|---|
| NS | PR8 | GFP | Caspase recognition site | Virus biology | [90] |
| NS | PR8 | GFP | Stop/start | Vaccine | [100] |
| NS | PR8 | maxGFP | 2A site | Virus pathogenesis | [71,101,102,103,104] |
| NS | PR8 | maxGFP, turboRFP, Gluc | 2A site | Antiviral and virus-host interaction | [48] |
| NS | PR8 pH1N1 | mCherry | 2A site | Antivirals, neutralizing antibodies, virus pathogenesis | [41] |
| NS | pH1N1 | Timer | 2A site | Virus propagation | [68] |
| NS | PR8 VN1203 | Venus, eGFP, eCFP, mCherry | 2A site | Virus-host interaction and virus pathogenesis | [38] |
| NS | PR8 WSN | GFP | 2× 2A site | Virus pathogenesis | [105,106] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breen, M.; Nogales, A.; Baker, S.F.; Martínez-Sobrido, L. Replication-Competent Influenza A Viruses Expressing Reporter Genes. Viruses 2016, 8, 179. https://doi.org/10.3390/v8070179
Breen M, Nogales A, Baker SF, Martínez-Sobrido L. Replication-Competent Influenza A Viruses Expressing Reporter Genes. Viruses. 2016; 8(7):179. https://doi.org/10.3390/v8070179
Chicago/Turabian StyleBreen, Michael, Aitor Nogales, Steven F. Baker, and Luis Martínez-Sobrido. 2016. "Replication-Competent Influenza A Viruses Expressing Reporter Genes" Viruses 8, no. 7: 179. https://doi.org/10.3390/v8070179
APA StyleBreen, M., Nogales, A., Baker, S. F., & Martínez-Sobrido, L. (2016). Replication-Competent Influenza A Viruses Expressing Reporter Genes. Viruses, 8(7), 179. https://doi.org/10.3390/v8070179