
Ectromelia Virus Disease Characterization in the BALB/c Mouse: A Surrogate Model for Assessment of Smallpox Medical Countermeasures

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Viral Material
2.3. Animals
2.4. Lethal Dose
2.5. Natural History of ECTV Infection Following IN Challenge of BALB/c Mice
2.5.1. Study Design
2.5.2. Body Weight and Temperature Collection
2.5.3. Clinical Observations
2.5.4. Viremia and Viral Titers
2.5.5. Hematology and Clinical Chemistry
2.5.6. Pathology
2.5.7. Statistical Analysis
3. Results
3.1. Determination of ECTV-Mos Lethal Dose by Intranasal Challenge
3.2. Natural History of Infection
3.2.1. Mortality Assessment
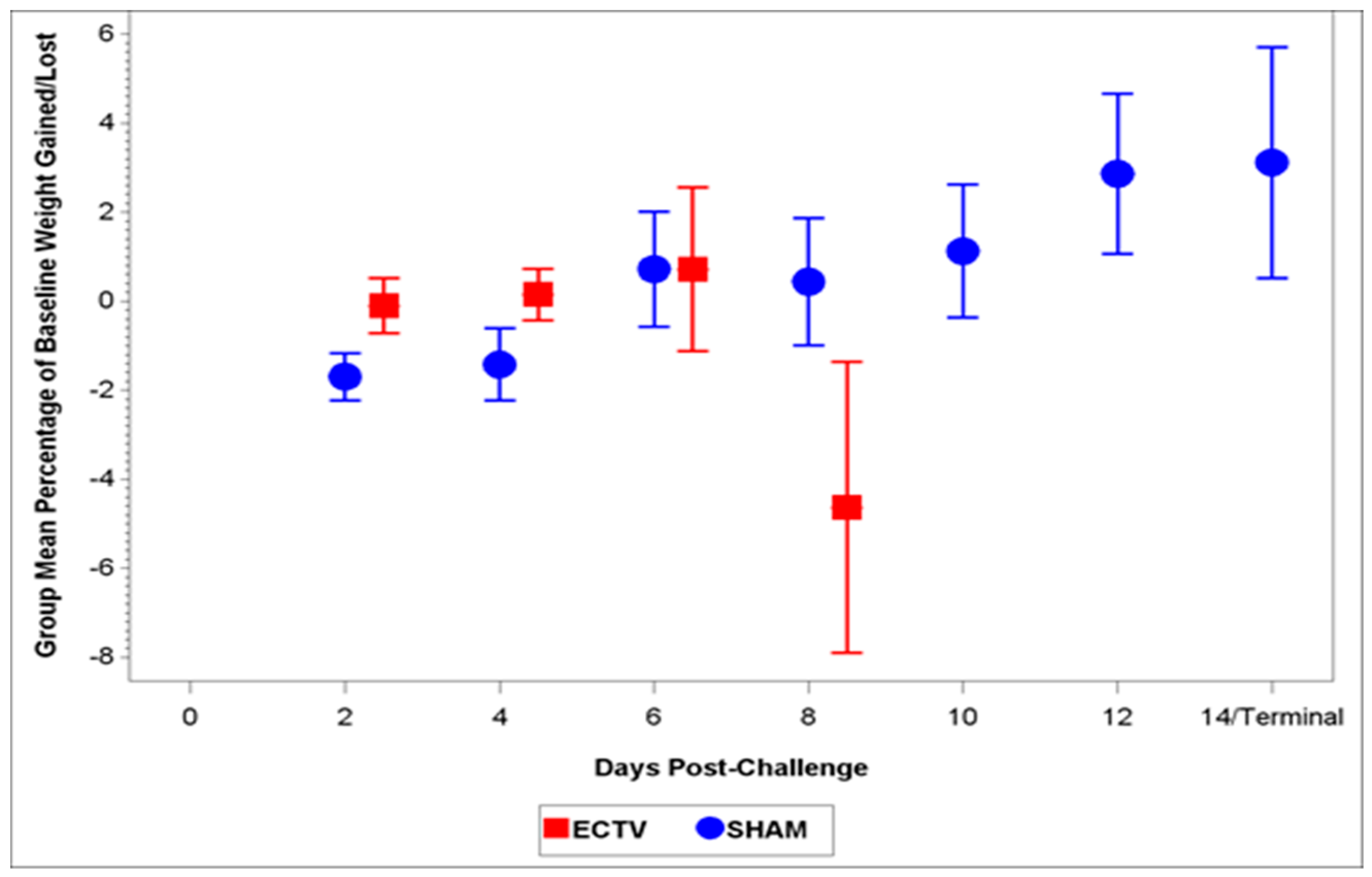
3.2.2. Body Weight
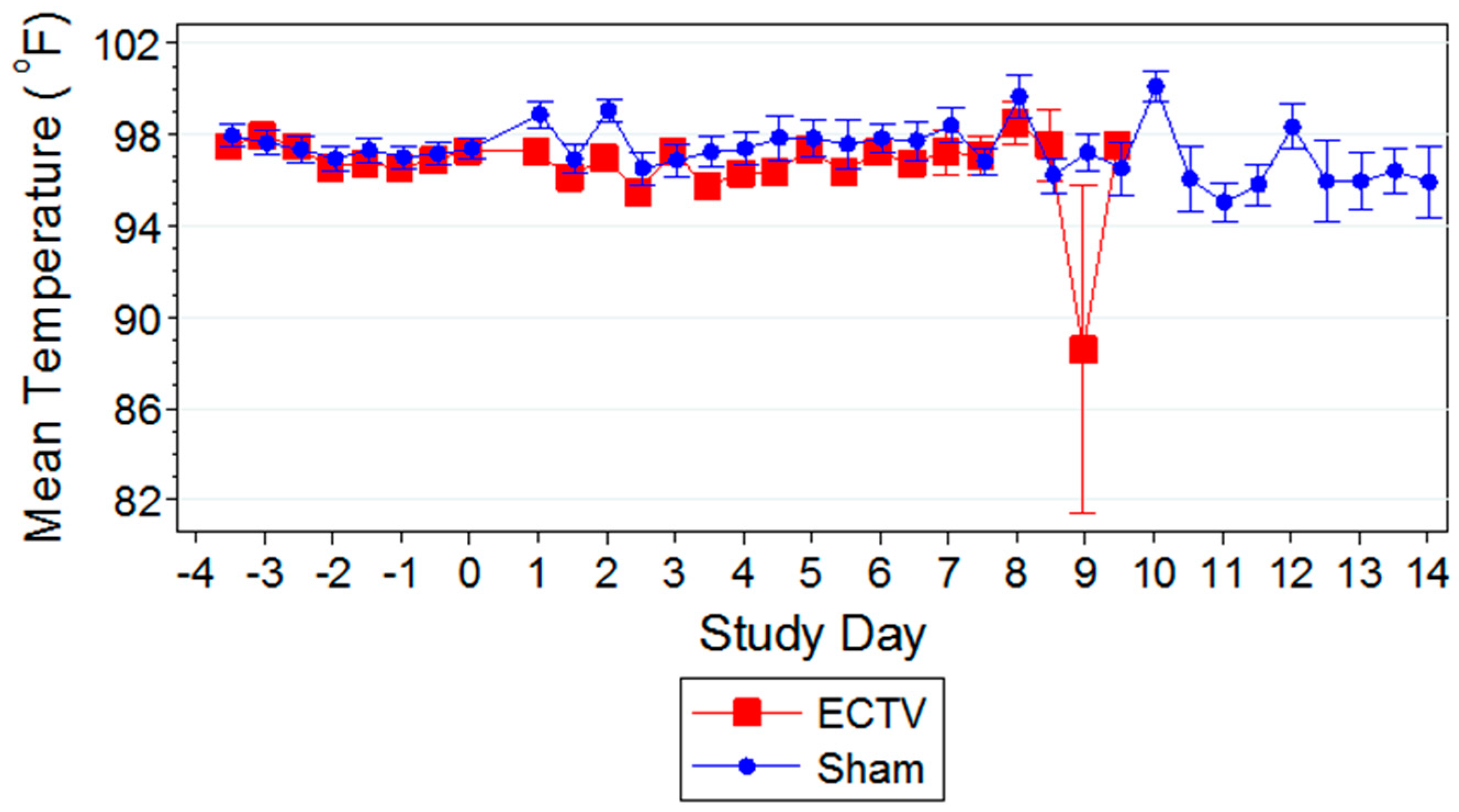
3.2.3. Body Temperature
3.2.4. Clinical Observations
3.3. ECTV Disease Progression
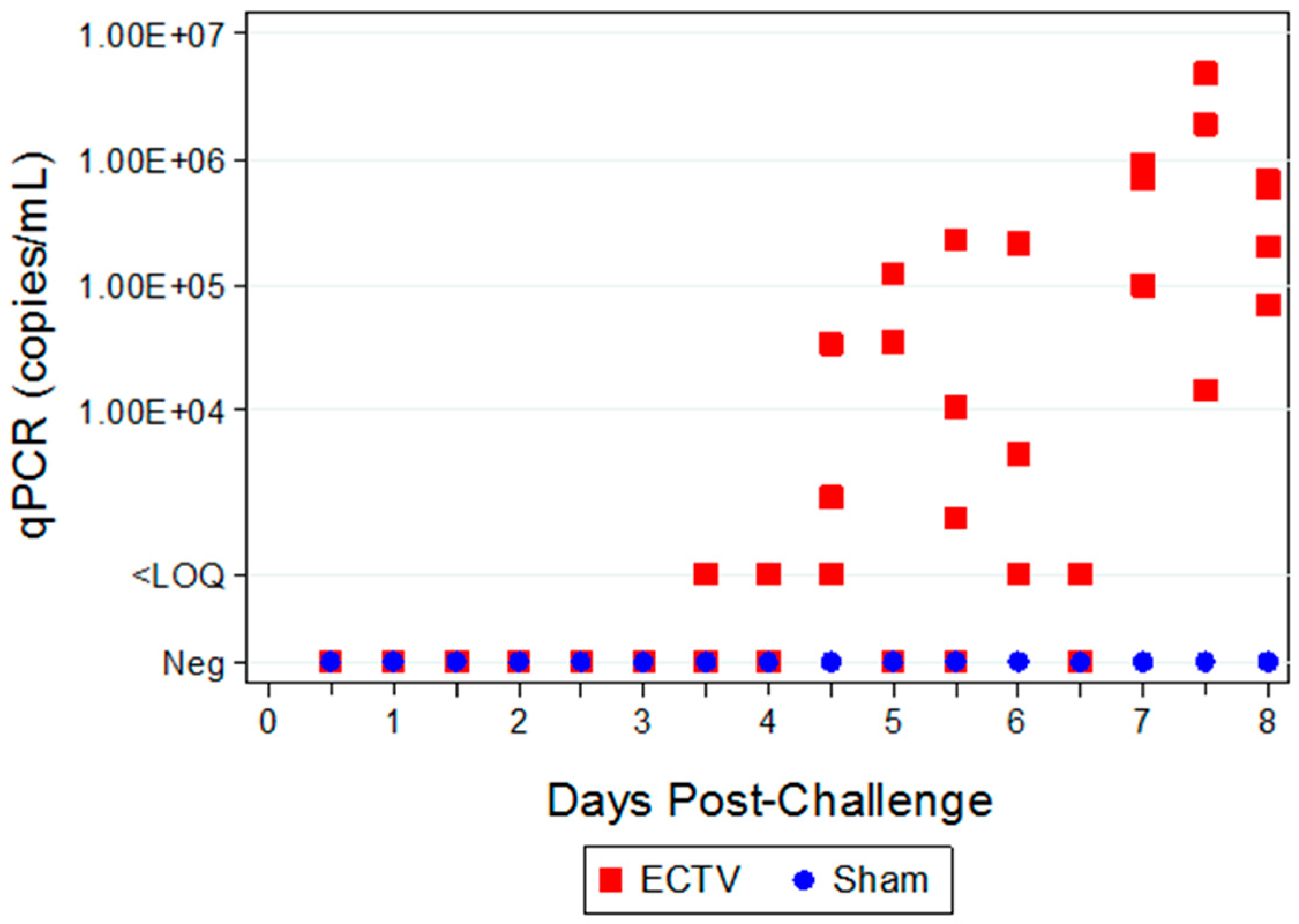
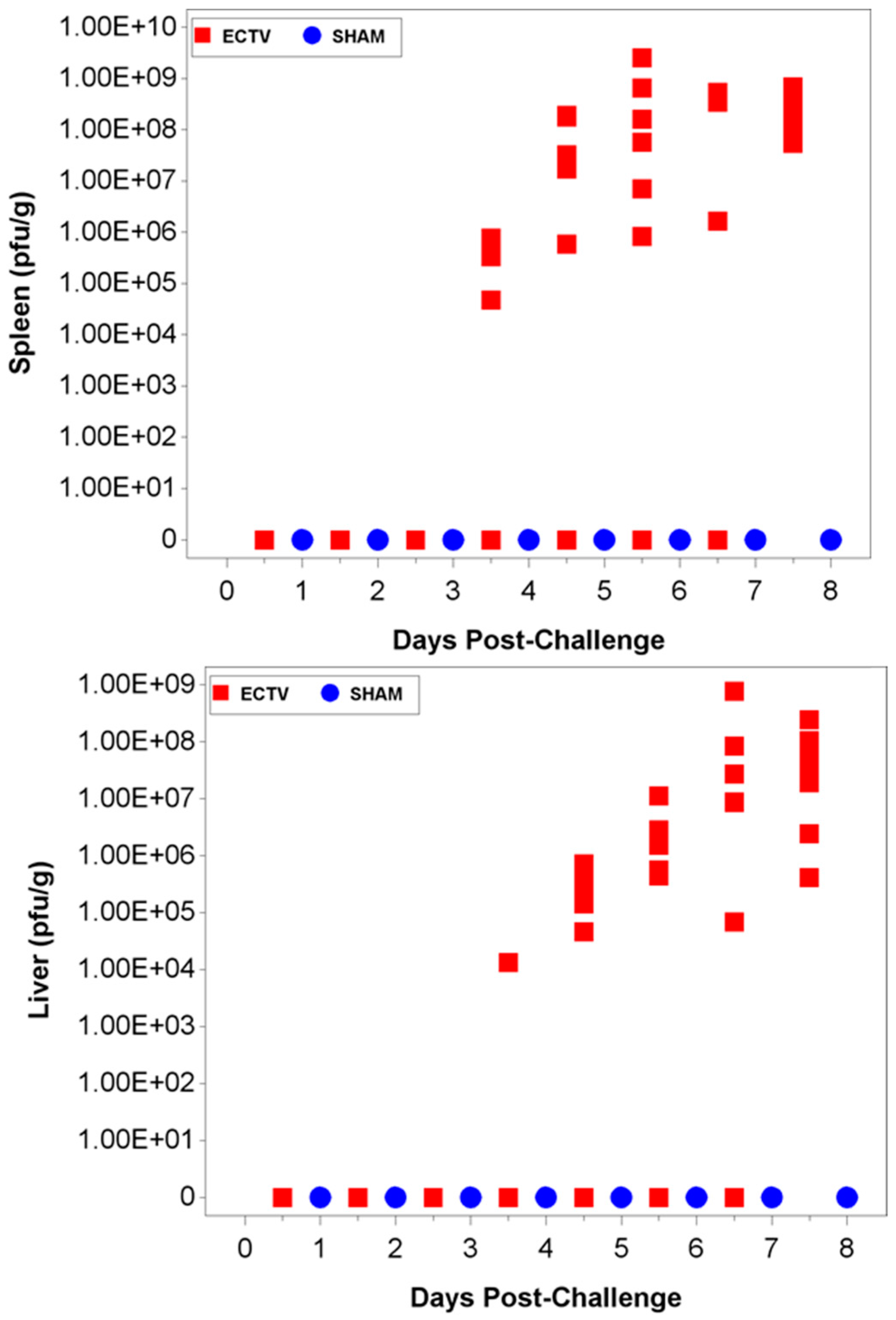
3.3.1. Viral Titers via qPCR
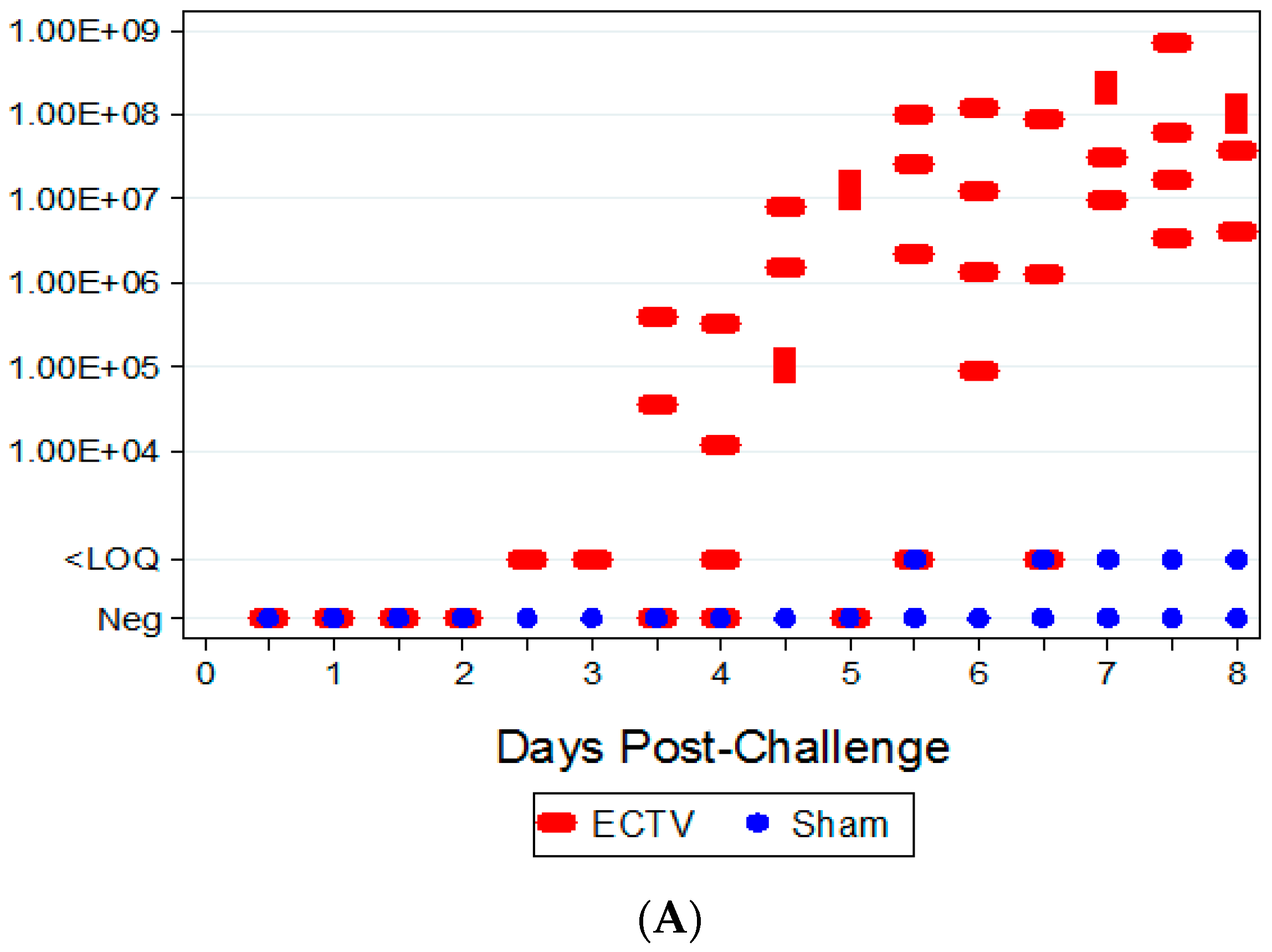
3.3.2. Viral Titers via Plaque Assay
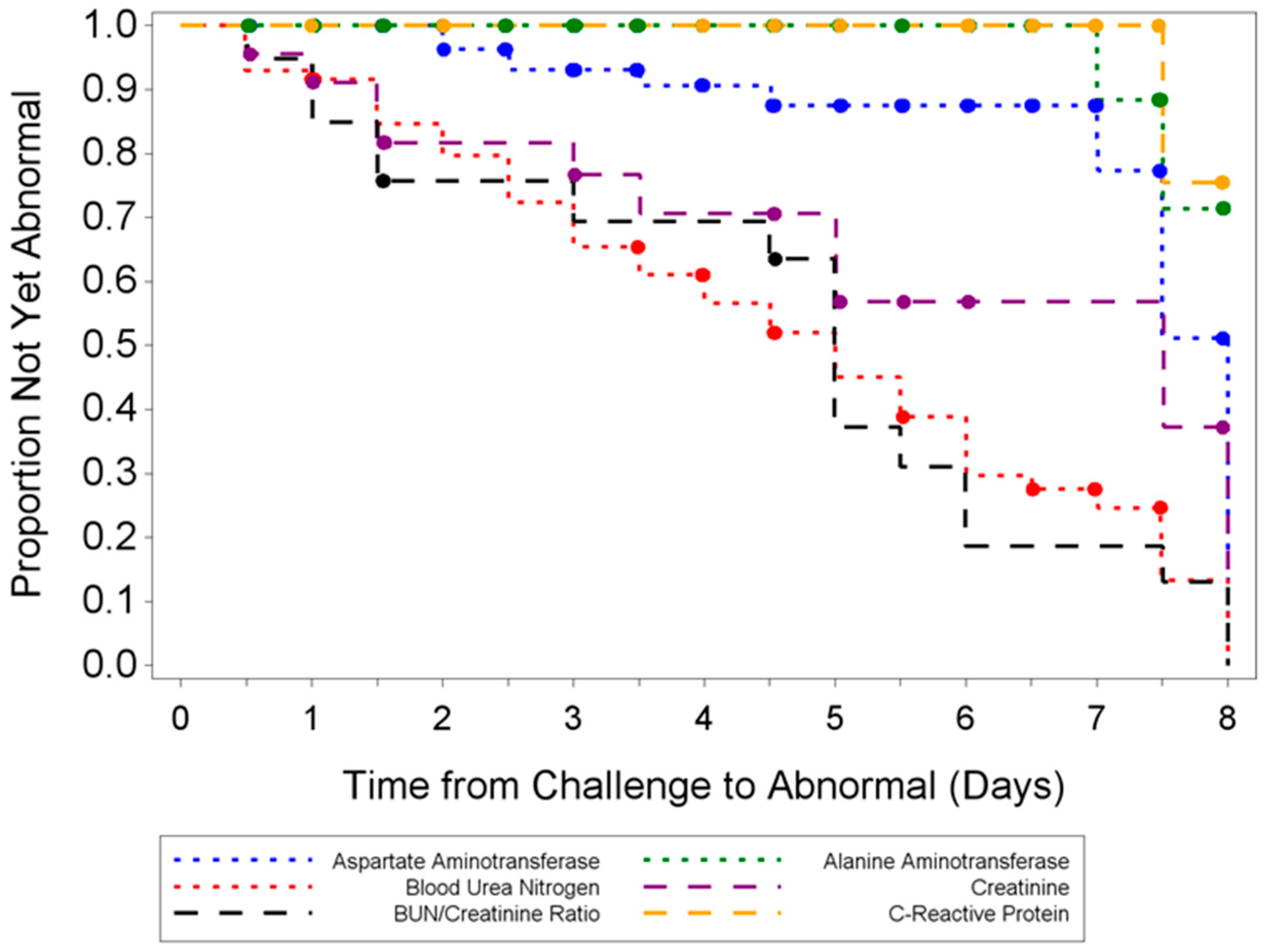
3.3.3. Hematology and Clinical Chemistry
3.3.4. Pathology
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D. Smallpox and its Eradication. In History of International and Public Health; World Health Organization: Geneva, Switzerland, 1988; Volume 6. [Google Scholar]
- Arita, I. Discovery of forgotten variola specimens at the National Institutes of Health in the USA. Expert Rev. Anti-Infect. Ther. 2014, 12, 1419–1421. [Google Scholar] [CrossRef] [PubMed]
- Nalca, A.; Zumbrun, E.E. ACAM2000TM: The new smallpox vaccine for United States Strategic National Stockpile. Drug Des. Devel. Ther. 2010, 4, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Grosenbach, D.W.; Jordan, R.; Hruby, D.E. Development of the small-molecule antiviral ST-246® as a smallpox therapeutic. Future Virol. 2011, 6, 653–671. [Google Scholar] [CrossRef] [PubMed]
- Marchal, J. Infectious ectromelia: A hitherto undescribed virus disease in mice. J. Pathol. Bacteriol. 1930, 33, 713–728. [Google Scholar] [CrossRef]
- Esteban, D.J.; Buller, R.M. Ectromelia virus: The causative agent of mousepox. J. Gen. Virol. 2005, 86, 2645–2659. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Ramsay, A.J.; Christensen, C.D.; Beaton, S.; Hall, D.F.; Ramshaw, I.A. Expression of Mouse Interleukin-4 by a Recombinant Ectromelia Virus Suppresses Cytolytic Lymphocyte Responses and Overcomes Genetic Resistance to Mousepox. J. Virol. 2001, 75, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.L.; Nichols, D.K.; Martinez, M.J.; Raymond, J.W. Animal models of orthopoxvirus infection. Vet. Pathol. 2010, 47, 852–870. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.; Siddiqui, A.M.; Painter, G.; Schriewer, J.; Buller, R.M. Ectromelia virus infections of mice as a model to support the licensure of anti-orthopoxvirus therapeutics. Viruses 2010, 2, 1918–1932. [Google Scholar] [CrossRef] [PubMed]
- Esteban, D.J.; Parker, S.; Schriewer, J.; Hartzler, H.; Buller, R.M. Mousepox, a small animal model of smallpox. Methods Mol. Biol. 2012, 890, 177–198. [Google Scholar] [PubMed]
- Buller, R.M. Mousepox: A small animal model for biodefense research. Appl. Biosaf. 2004, 9, 10–19. [Google Scholar] [CrossRef]
- O’Neill, H.C.; Brenan, M. A role for early cytotoxic T cells in resistance to ectromelia virus infection in mice. J. Gen. Virol. 1987, 68, 2669–2673. [Google Scholar] [CrossRef] [PubMed]
- Percy, D.H.; Barthold, S.W. Mouse. Pathology of Laboratory Rodents and Rabbits, 3rd ed.; Blackwell Publishing: Ames, IA, USA, 2007; pp. 3–124. [Google Scholar]
- Smee, D.F.; Sidwell, R.W. A review of compounds exhibiting anti-orthopoxvirus activity in animal models. Antivir. Res. 2003, 57, 41–52. [Google Scholar] [CrossRef]
- Buller, R.M. The BALB/c mouse as a model to study orthopoxviruses. Curr. Top. Microbiol. Immunol. 1985, 122, 148–153. [Google Scholar] [PubMed]
- Smallwood, S.E.; Rahman, M.M.; Smith, D.W.; McFadden, G. Myxoma Virus: Propagation, Purification, Quantification, and Storage. Curr. Protoc. Microbiol. 2010, 14A.1. [Google Scholar] [CrossRef]
- Parker, S.; Chen, N.G.; Foster, S.; Hartzler, H.; Hembrador, E.; Hruby, D.; Jordan, R.; Lanier, R.; Painter, G.; Painter, W.; et al. Evaluation of disease and viral biomarkers as triggers for therapeutic intervention in respiratory mousepox—An animal model of smallpox. Antivir. Res. 2012, 94, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Kulesh, D.A.; Baker, R.O.; Loveless, B.M.; Norwood, D.; Zwiers, S.H.; Mucker, E.; Hartmann, C.; Herrera, R.; Miller, D.; Christensen, D.; et al. Smallpox and pan-orthopox virus detection by real-time 3′-minor groove binder TaqMan assays on the Roche LightCycler and the Cepheid Smart Cycler platforms. J. Clin. Microbiol. 2004, 42, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Aldhamen, Y.A.; Seregin, S.S.; Kousa, Y.A.; Rastall, D.P.; Appledorn, D.M.; Godbehere, S.; Schutte, B.C.; Amalfitano, A. Improved cytotoxic T-lymphocyte immune responses to a tumor antigen by vaccines co-expressing the SLAM-associated adaptor EAT-2. Cancer Gene Ther. 2013, 20, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA) Center for Drug Evaluation and Research (CDER). Antiviral Drugs Advisory Committee Meeting, Silver Spring, MD, USA, 14–15 December 2011.
- Johnson, R.F.; Dyall, J.; Ragland, D.R.; Huzella, L.; Byrum, R.; Jett, C.; St. Claire, M.; Smith, A.L.; Paragas, J.; Blaney, J.E.; et al. Comparative Analysis of Monkeypox Virus Infection of Cynomologus Macaques by the Intravenous or Intrabronchial Inoculation Route. J. Virol. 2011, 85, 2112–2125. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F.; Buller, R.M. Mousepox. In Viral Pathogenesis; Lippincott-Raven Publishing: Philadelphia, PA, USA, 1997; pp. 535–553. [Google Scholar]
- Scalzo, A.A.; Farrell, H.E.; Karupiah, G. Techniques for studying murine natural killer cells in defense against viral infection. Methods Mol. Biol. 2000, 121, 163–177. [Google Scholar] [PubMed]
- Fang, M.; Siciliano, N.A.; Hersperger, A.R.; Roscoe, F.; Hu, A.; Ma, X.; Shamsedeen, A.R.; Eisenlohr, L.C.; Sigal, L.J. Perforin-dependent CD4+ T-cell cytotoxicity contributes to control a murine poxvirus infection. Proc. Natl. Acad. Sci. USA 2012, 109, 9983–9988. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.; Karupiah, G.; Takeda, K.; Akira, S.; Matthaei, K.I.; Foster, P.S. Enhanced resistance in STAT6-deficient mice to infection with ectromelia virus. Proc. Natl. Acad. Sci. USA 2001, 98, 6812–6817. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Inoculation | No. of Animals (M/F) | Oropharyngeal Secretions Collection Times - Swabs (Hours PI) | qPCR/Plaque Assay Collection Times (Hours) | Hematology/Clinical Chemistry Collection Times (Hours PI) |
|---|---|---|---|---|---|
| 1 | ECTV | 4/4 | 12, 24 | 12, 24 | NA |
| 2 | SHAM | 2/2 | |||
| 3 | ECTV | 4/4 | 36, 48 | 36, 48 | |
| 4 | SHAM | 2/2 | |||
| 5 | ECTV | 4/4 | 60, 72 | 60, 72 | |
| 6 | SHAM | 2/2 | |||
| 7 | ECTV | 4/4 | 84, 96 | 84, 96 | |
| 8 | SHAM | 2/2 | |||
| 9 | ECTV | 4/4 | 108, 120 | 108, 120 | |
| 10 | SHAM | 2/2 | |||
| 11 | ECTV | 4/4 | 132, 144 | 132, 144 | |
| 12 | SHAM | 2/2 | |||
| 13 | ECTV | 4/4 | 156, 168 | 156, 168 | |
| 14 | SHAM | 2/2 | |||
| 15 | ECTV | 4/4 | 180, 192 | 180, 192 | |
| 16 | SHAM | 2/2 | |||
| 17 | ECTV | 2/2 | NA | NA | 0 |
| 18 | ECTV | 2/2 | 12 | ||
| 19 | ECTV | 2/2 | 24 | ||
| 20 | ECTV | 2/2 | 36 | ||
| 21 | ECTV | 2/2 | 48 | ||
| 22 | ECTV | 2/2 | 60 | ||
| 23 | ECTV | 2/2 | 72 | ||
| 24 | ECTV | 2/2 | 84 | ||
| 25 | ECTV | 2/2 | 96 | ||
| 26 | ECTV | 2/2 | 108 | ||
| 27 | ECTV | 2/2 | 120 | ||
| 28 | ECTV | 2/2 | 132 | ||
| 29 | ECTV | 2/2 | 144 | ||
| 30 | ECTV | 2/2 | 156 | ||
| 31 | ECTV | 2/2 | 168 | ||
| 32 | ECTV | 2/2 | 180 | ||
| 33 | ECTV | 2/2 | 192 | ||
| 34 | ECTV | 4/4 | NA | ||
| 35 | SHAM | 4/4 |
| Severity of Condition | Clinical Condition/Definition |
|---|---|
| General Appearance | |
| Mild (1) | 1 of 3 of the following signs: Lethargy, Hunched Posture, Ruffled Fur |
| Moderate (2) | 2 of 3 of the following signs: Lethargy, Hunched Posture, Ruffled Fur |
| Severe (3) | 3 of 3 of the following signs: Lethargy, Hunched Posture, Ruffled Fur or Lesions present |
| Dehydration | |
| Mild (1) | Slight decrease in skin turgor, skin will not tent |
| Moderate (2) | Moderate decrease in skin turgor; skin will tent and slowly (several seconds) return to normal |
| Severe (3) | Skin visibly wrinkled, dry; greatly decreased skin turgor, skin will tent |
| Dyspnea | |
| Mild (1) | Undefined |
| Moderate (2) | Demonstrates respiratory abnormalities |
| Severe (3) | Demonstrates labored breathing |
| Ocular Abnormalities | |
| Mild (1) | Clear discharge from one or both eyes |
| Moderate (2) | Clear discharge from one or both eye; partial closure of one eye |
| Severe (3) | Clear discharge from one or both eyes; and/or partial closure of both eyes |
| Group | Target Dose (PFU) | Back Titer (PFU) | Number of Animals Succumbed/Total Number of Animals | Mortality Proportion (Binomial Exact 95% Confidence Interval) |
|---|---|---|---|---|
| 1 | 5 | 2.44 | 10/16 | 0.63 (0.35, 0.85) |
| 2 | 50 | 27.50 | 14/16 | 0.88 (0.62, 0.98) |
| 3 | 200 | 119.50 | 15/16 | 0.94 (0.70, 1.00) |
| 4 | 500 | 292.50 | 16/16 | 1.00 (0.79, 1.00) |
| 5 | 1000 | 700 | 16/16 | 1.00 (0.79, 1.00) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garver, J.; Weber, L.; Vela, E.M.; Anderson, M.; Warren, R.; Merchlinsky, M.; Houchens, C.; Rogers, J.V. Ectromelia Virus Disease Characterization in the BALB/c Mouse: A Surrogate Model for Assessment of Smallpox Medical Countermeasures. Viruses 2016, 8, 203. https://doi.org/10.3390/v8070203
Garver J, Weber L, Vela EM, Anderson M, Warren R, Merchlinsky M, Houchens C, Rogers JV. Ectromelia Virus Disease Characterization in the BALB/c Mouse: A Surrogate Model for Assessment of Smallpox Medical Countermeasures. Viruses. 2016; 8(7):203. https://doi.org/10.3390/v8070203
Chicago/Turabian StyleGarver, Jennifer, Lauren Weber, Eric M. Vela, Mike Anderson, Richard Warren, Michael Merchlinsky, Christopher Houchens, and James V. Rogers. 2016. "Ectromelia Virus Disease Characterization in the BALB/c Mouse: A Surrogate Model for Assessment of Smallpox Medical Countermeasures" Viruses 8, no. 7: 203. https://doi.org/10.3390/v8070203
APA StyleGarver, J., Weber, L., Vela, E. M., Anderson, M., Warren, R., Merchlinsky, M., Houchens, C., & Rogers, J. V. (2016). Ectromelia Virus Disease Characterization in the BALB/c Mouse: A Surrogate Model for Assessment of Smallpox Medical Countermeasures. Viruses, 8(7), 203. https://doi.org/10.3390/v8070203