Oncolytic Reovirus Infection Is Facilitated by the Autophagic Machinery



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Buffers
2.2. Cell Lines
2.3. Viruses
2.4. Yield Quantification
2.5. Western Blotting
2.6. eGFP-LC3 Fluorescence Microscopy
2.7. Acridine Orange Staining
2.8. Electron Microscopy
3. Results
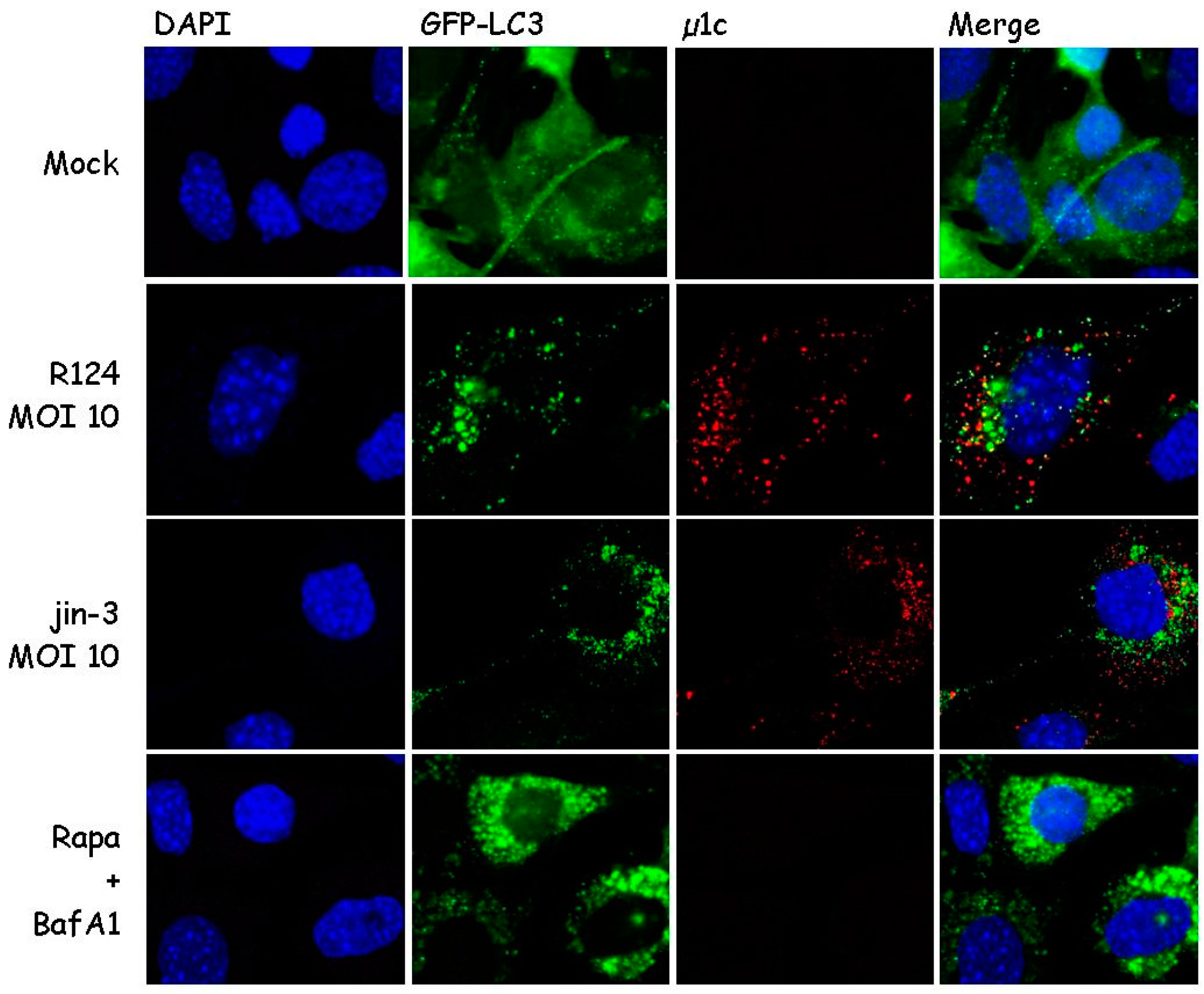
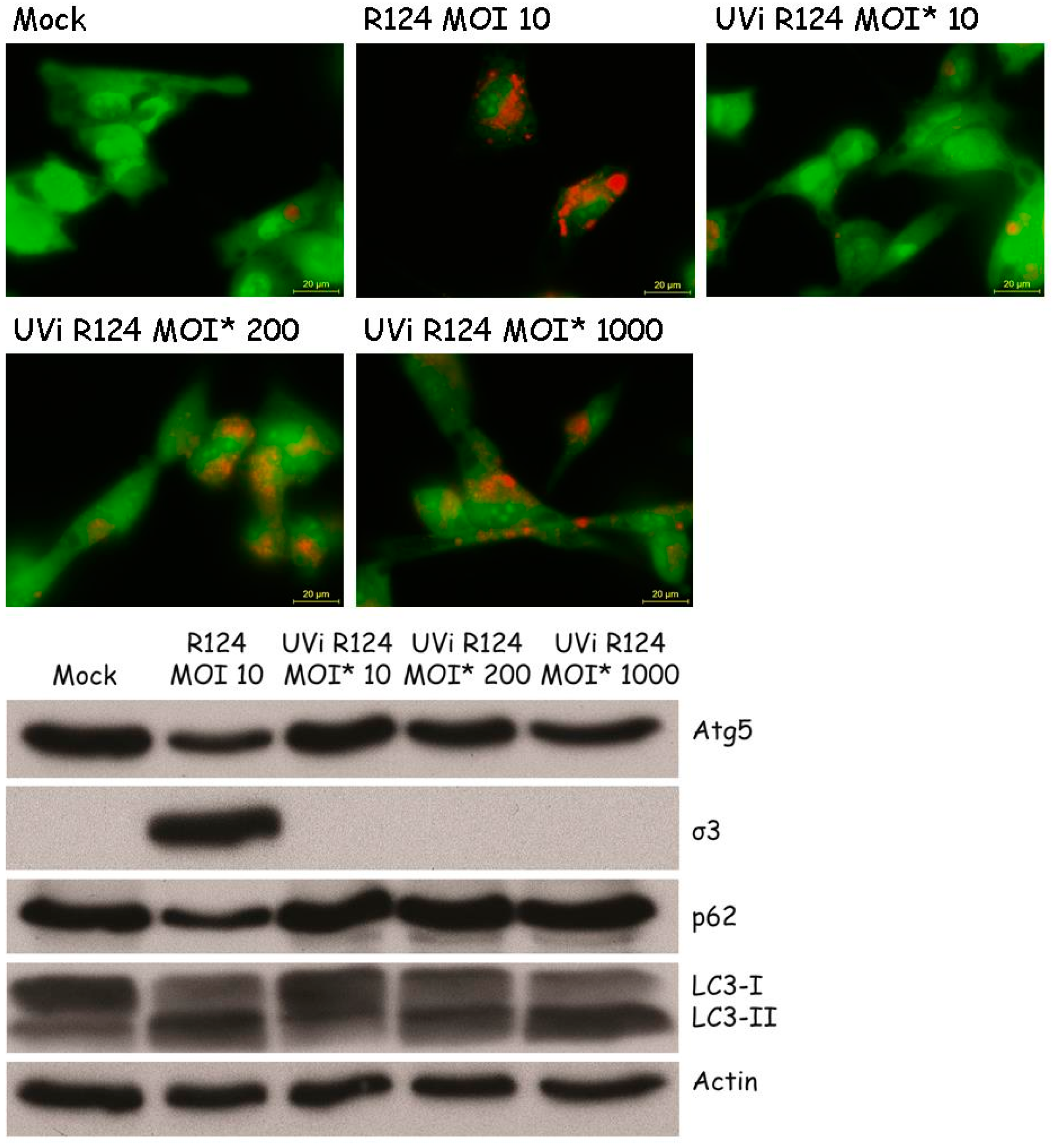
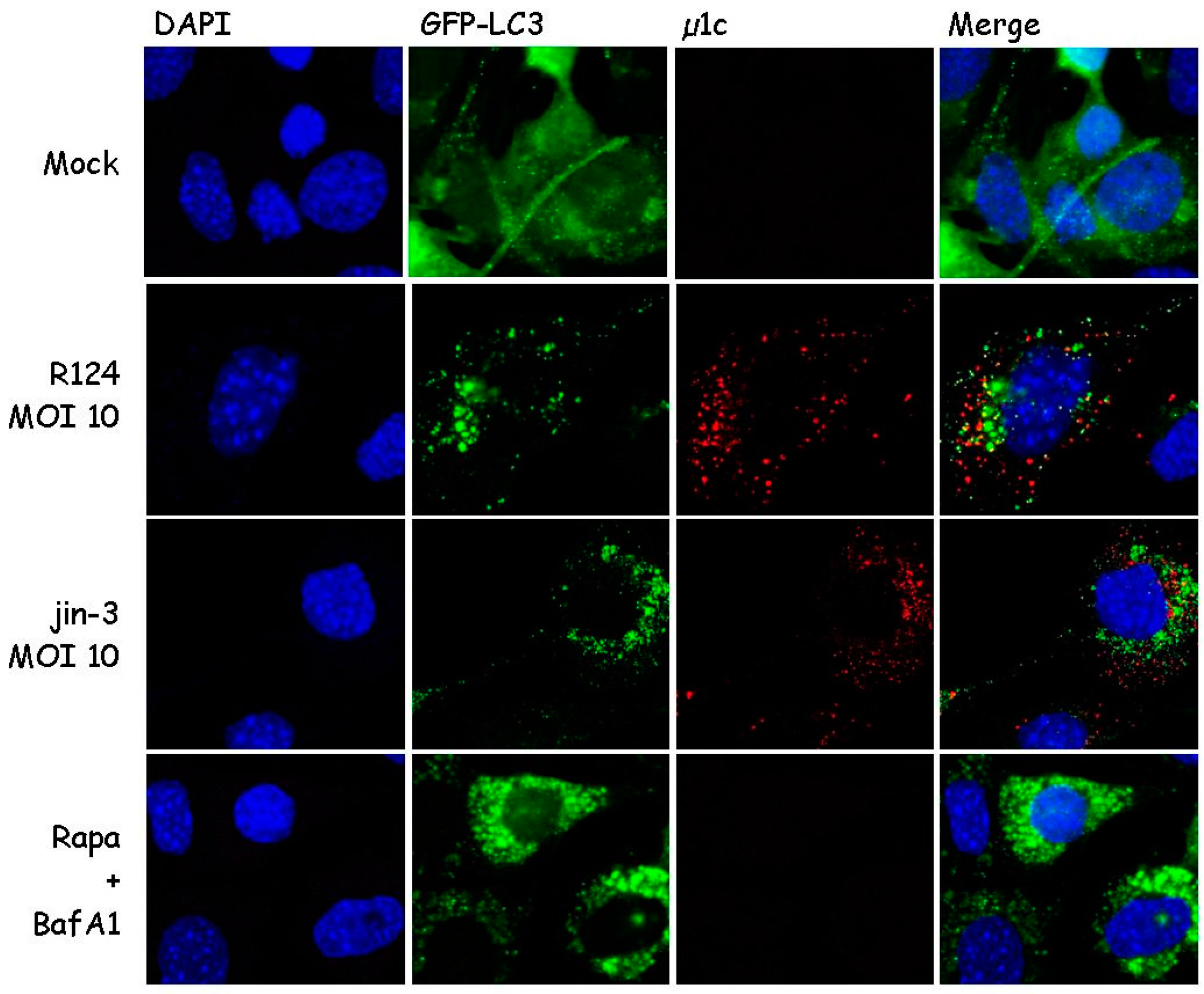
3.1. Reovirus Induces LC3 Puncta in MEFs
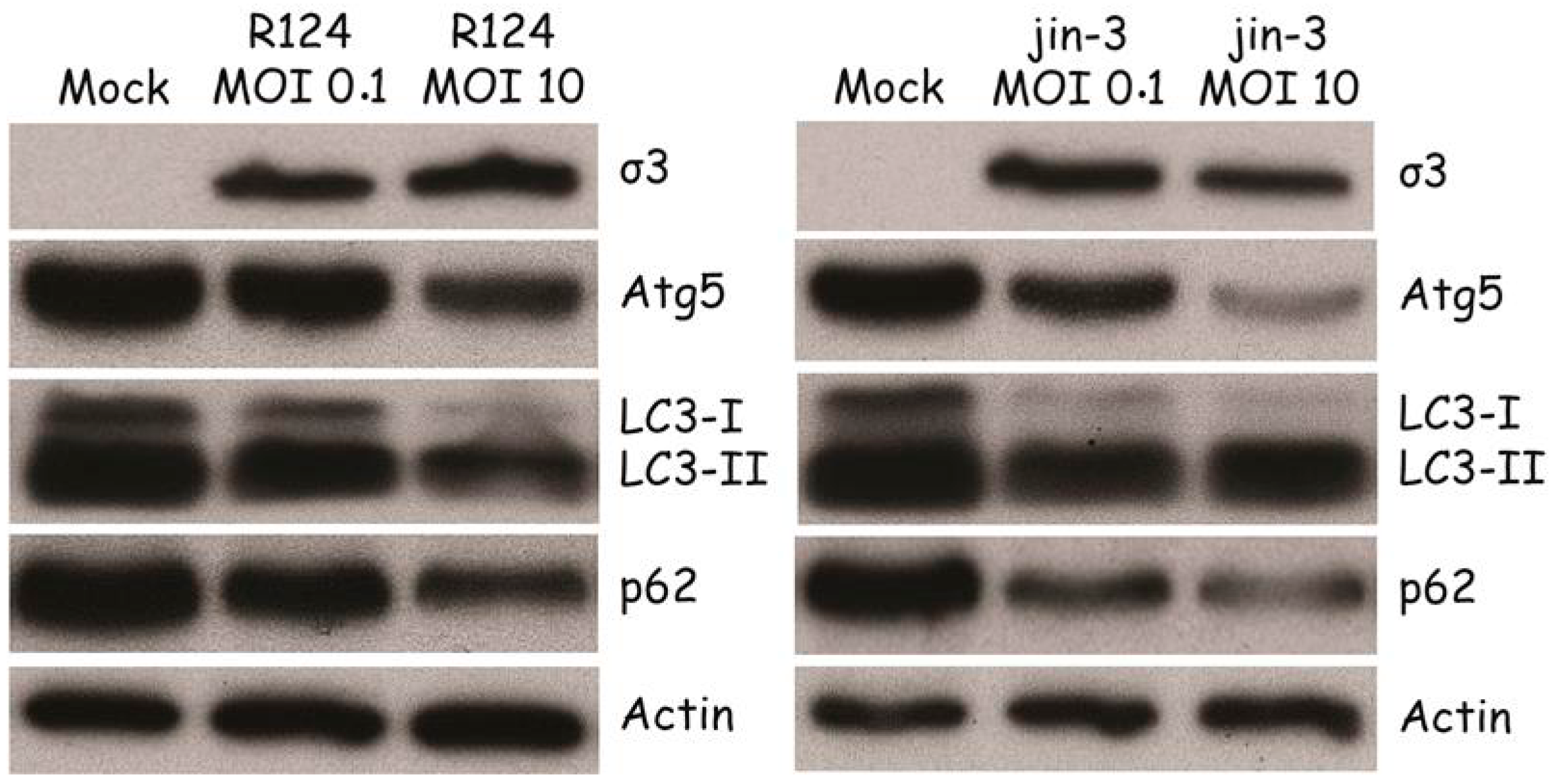
3.2. Reovirus Induces Atg5-Atg12 Conjugation, p62 Degradation, and LC3 Lipidation in MEFs
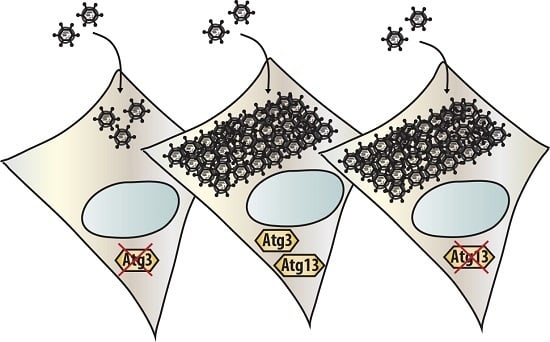
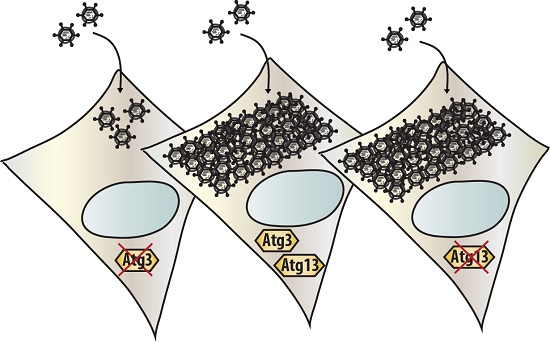
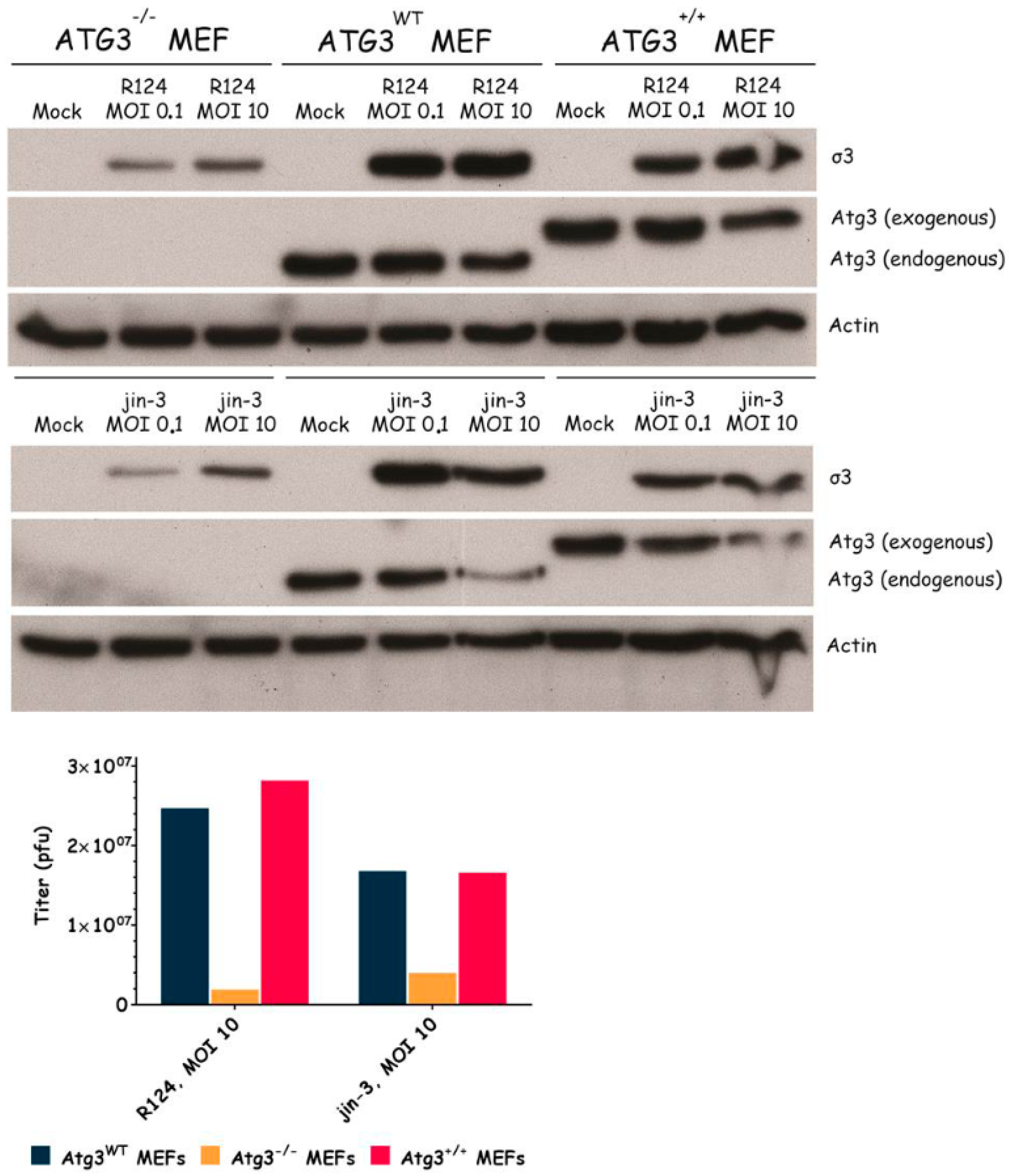
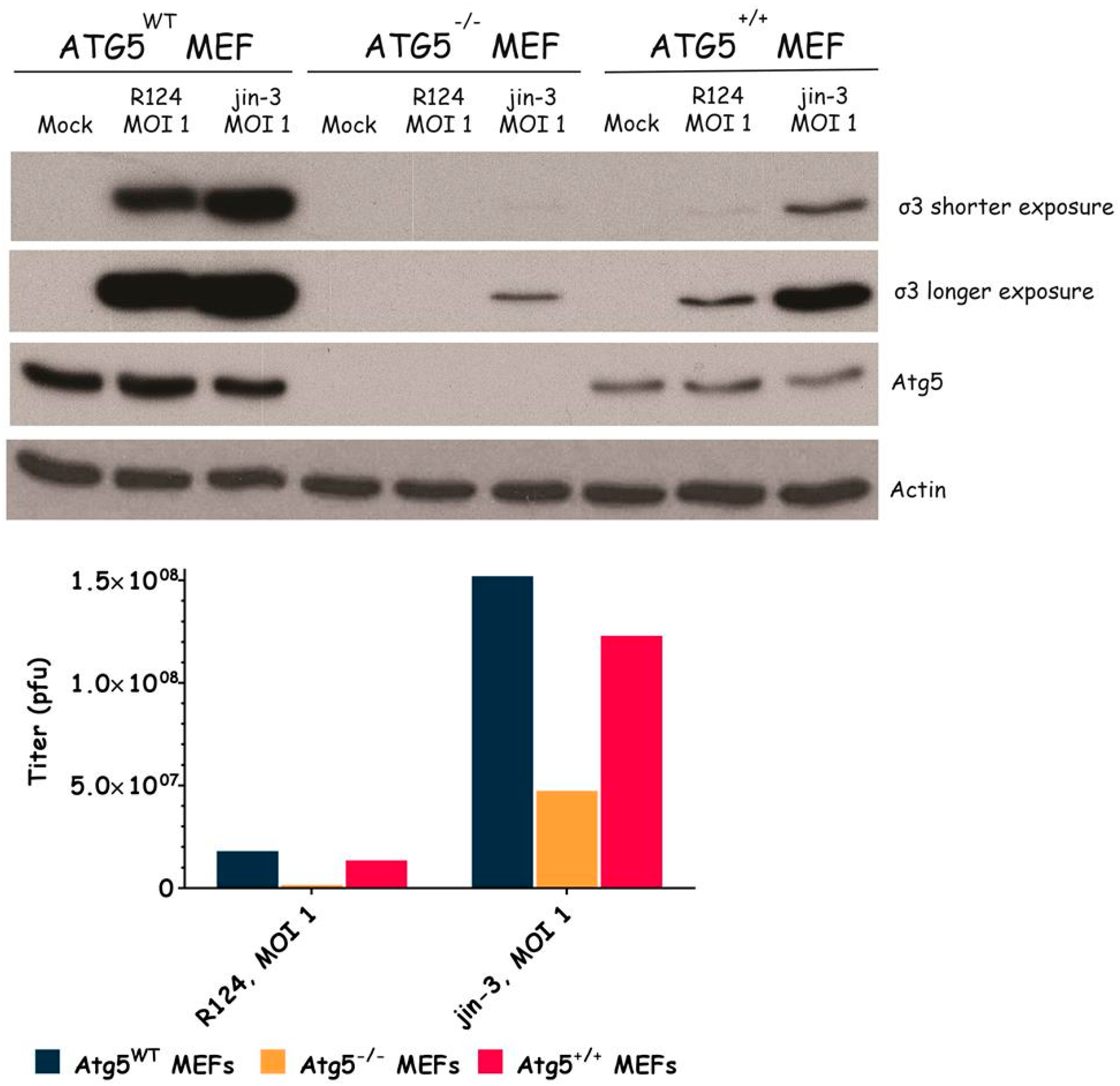
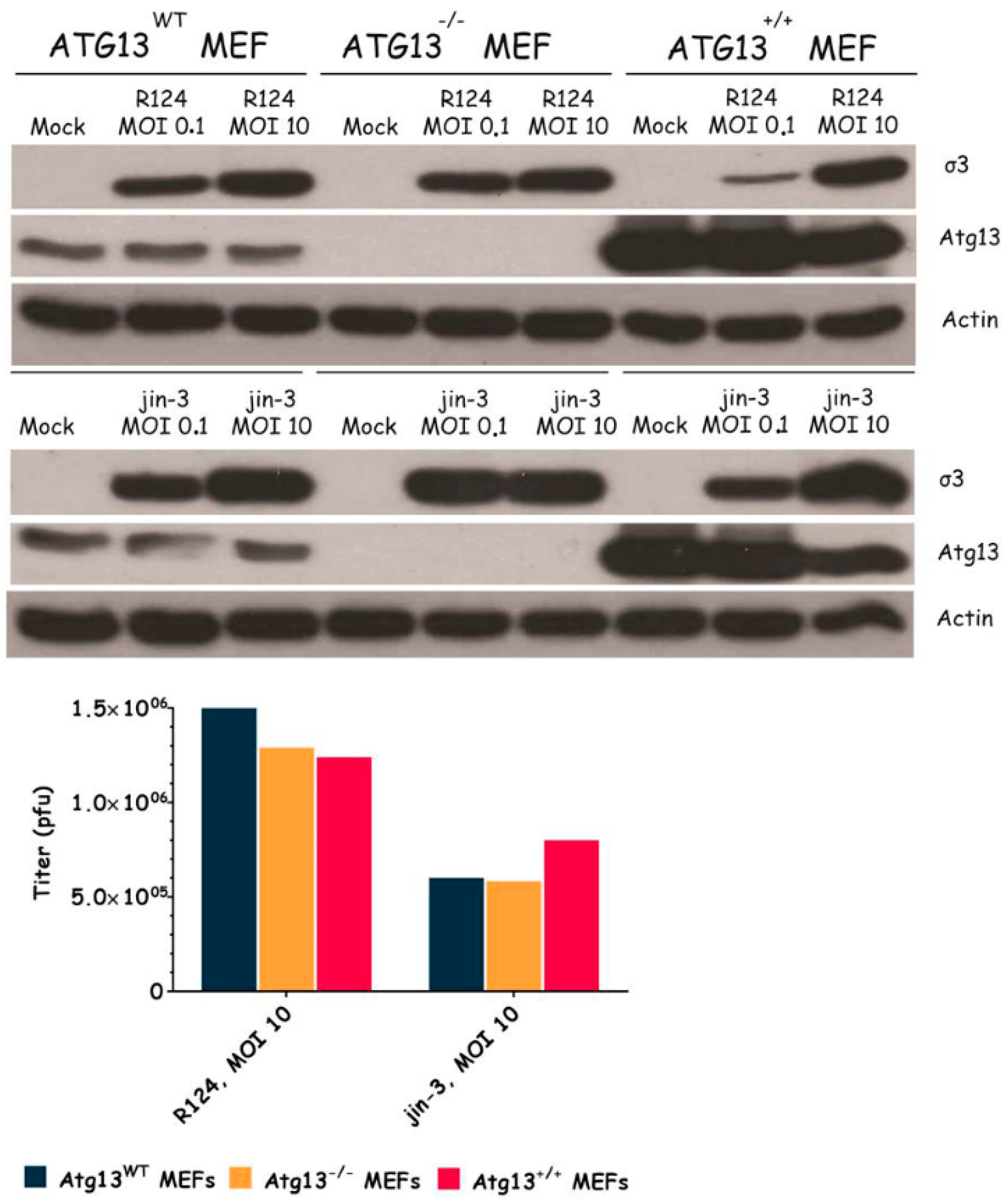
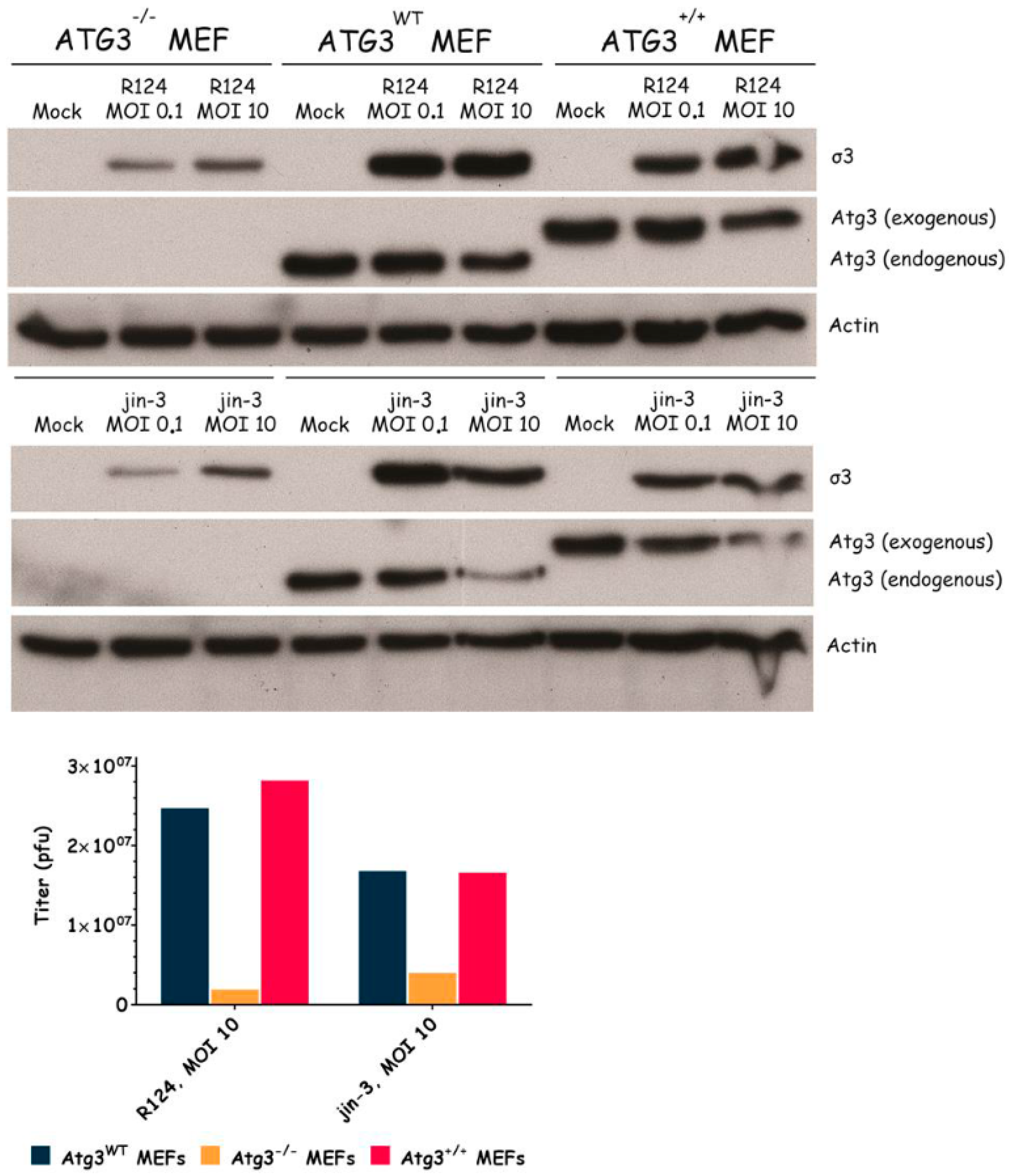
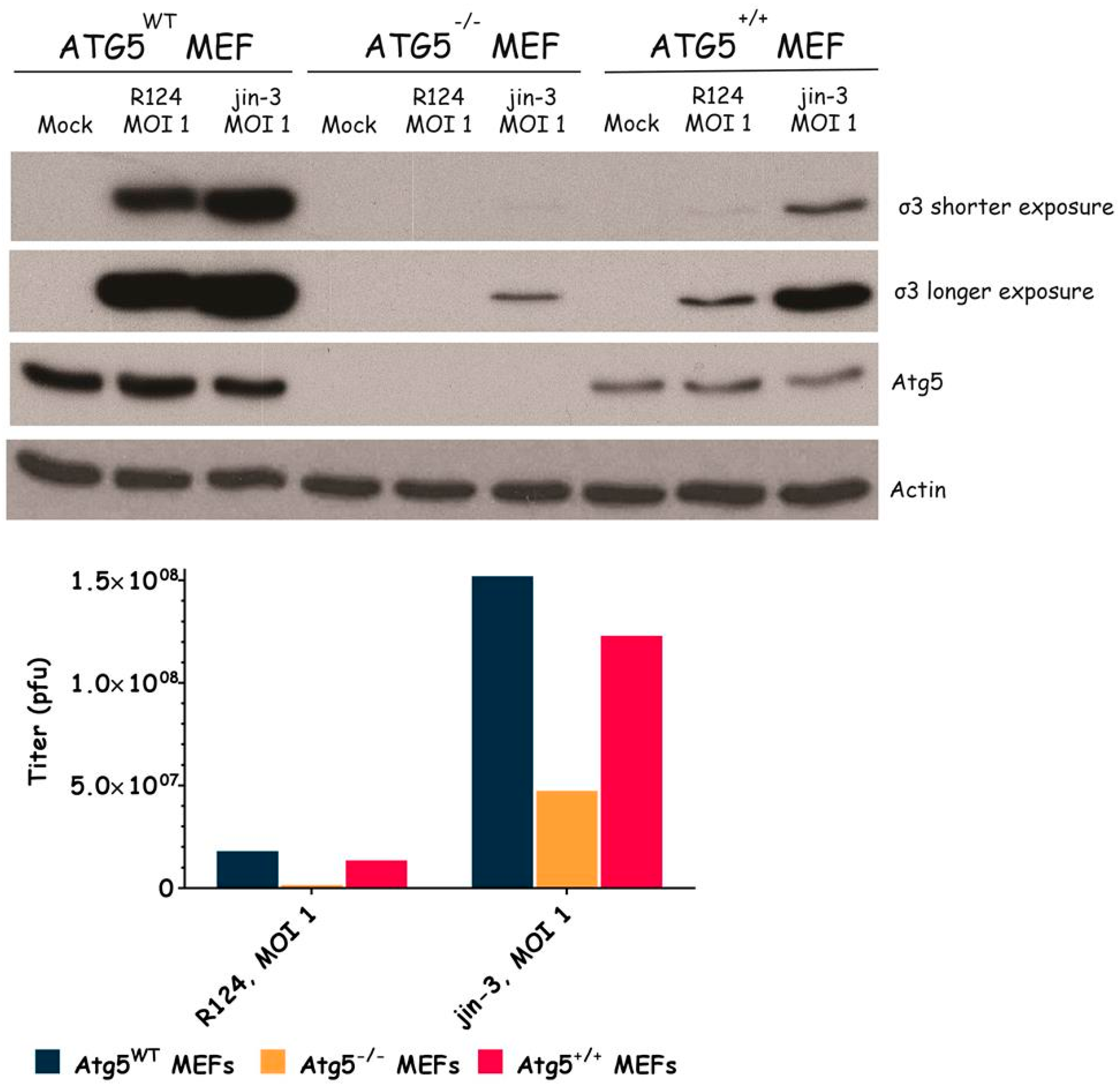
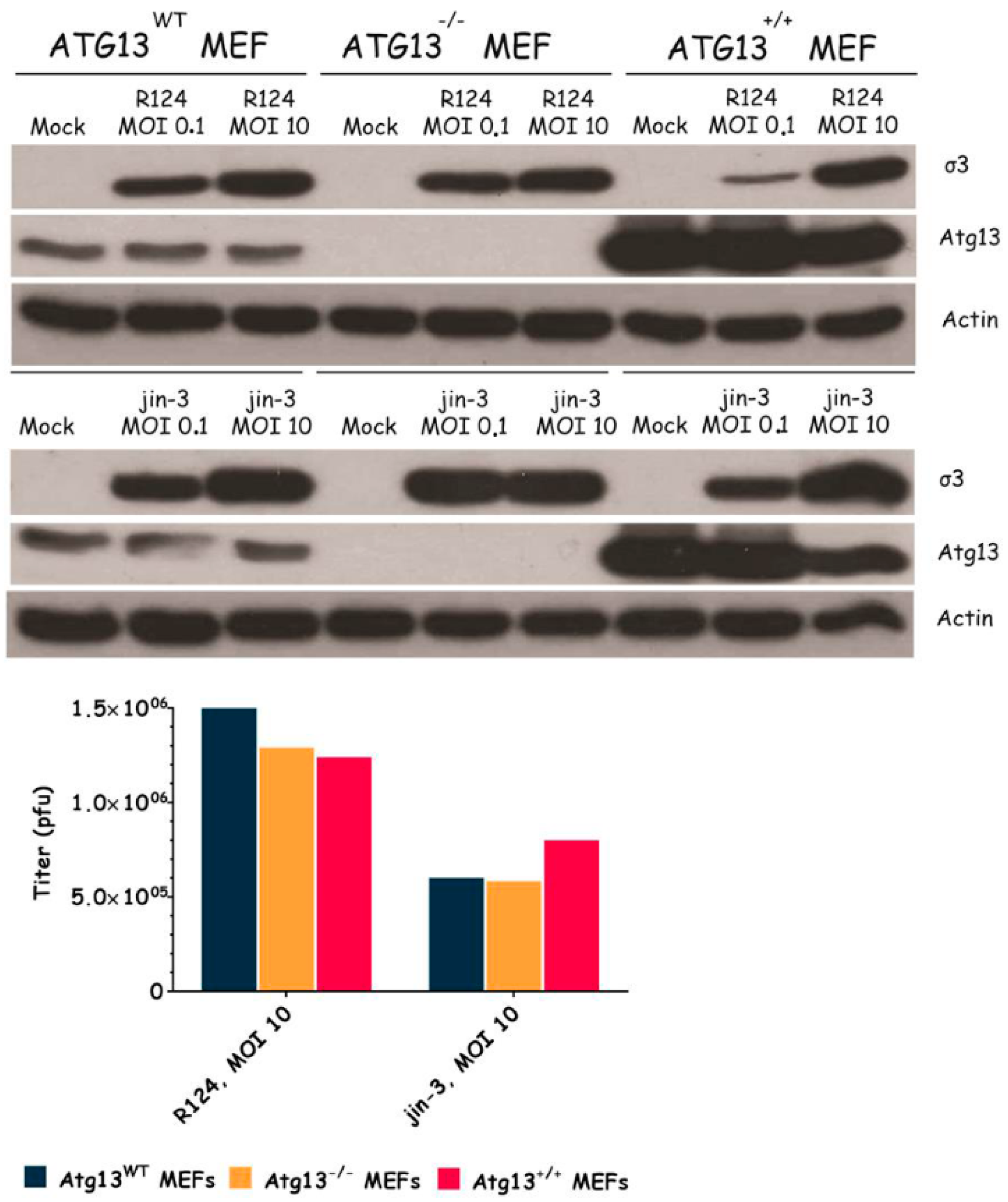
3.3. Atg3 and Atg5 but Not Atg13 Expression Facilitate Reovirus Replication
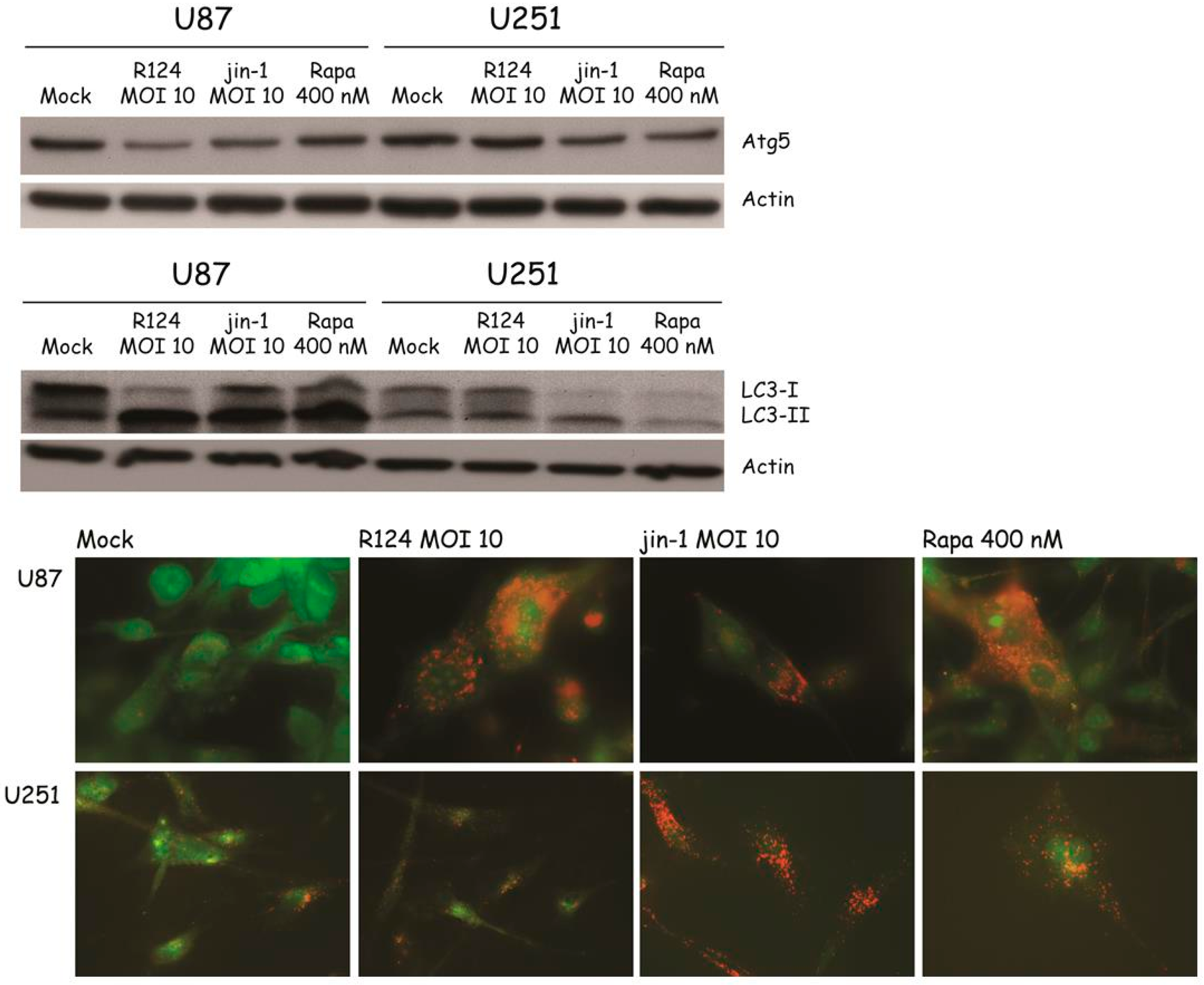
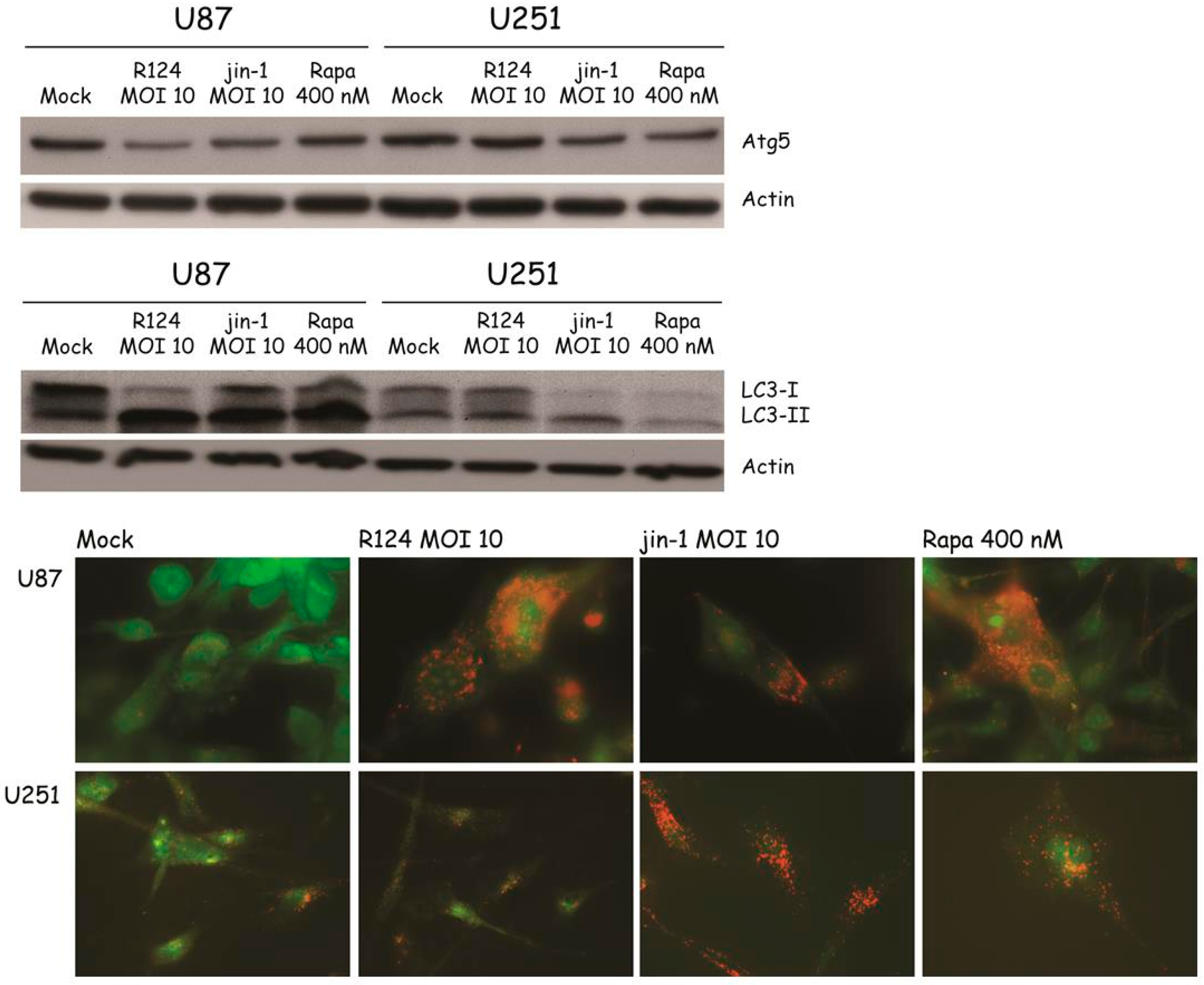
3.4. Reovirus Induces Atg5-Atg12 Conjugation, LC3 Lipidation, and Acidic Vesicular Organelles in Glioblastoma Cell Lines
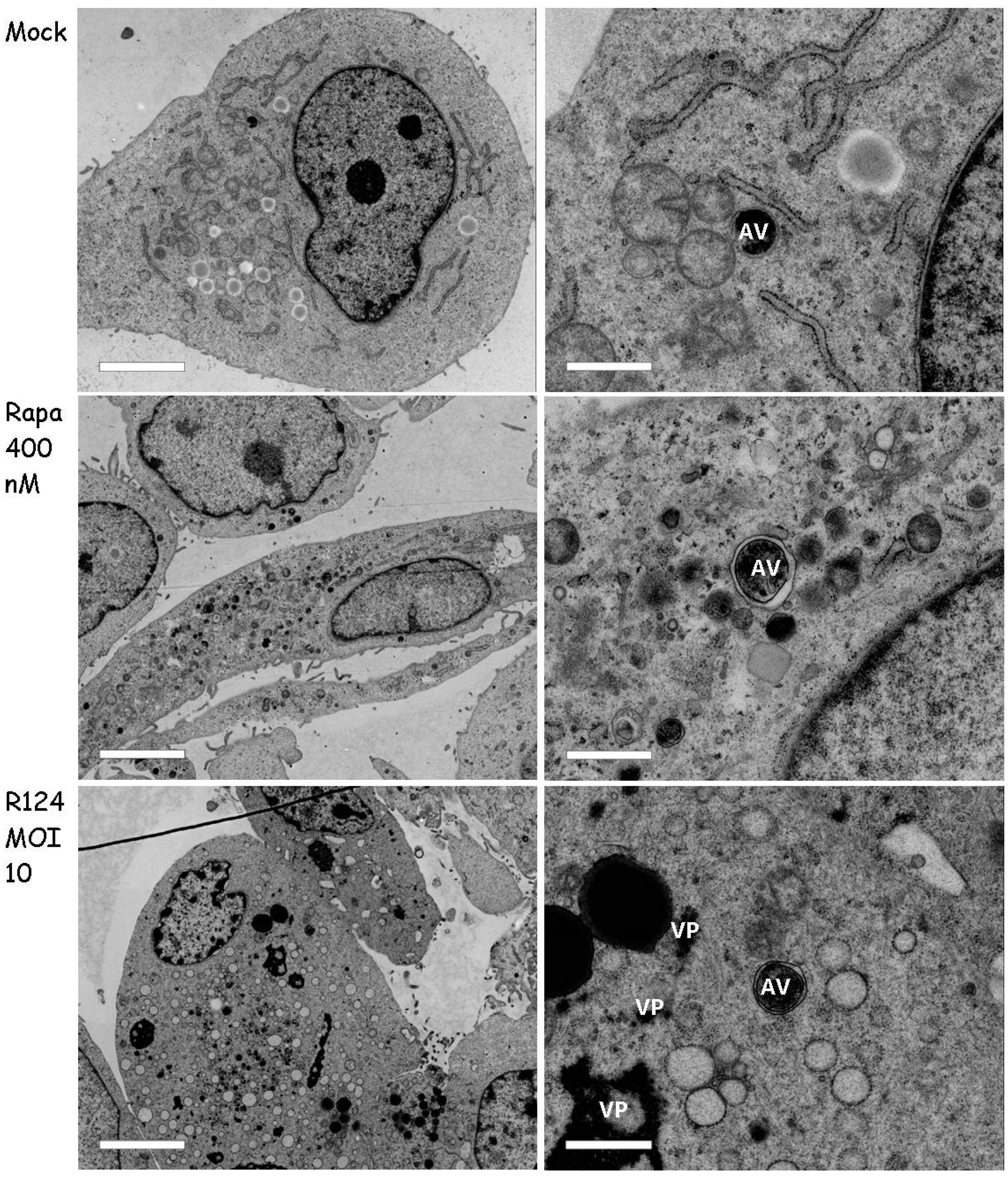
3.5. Autophagosomes Can Be Observed in Reovirus-Infected U87-MG Cells
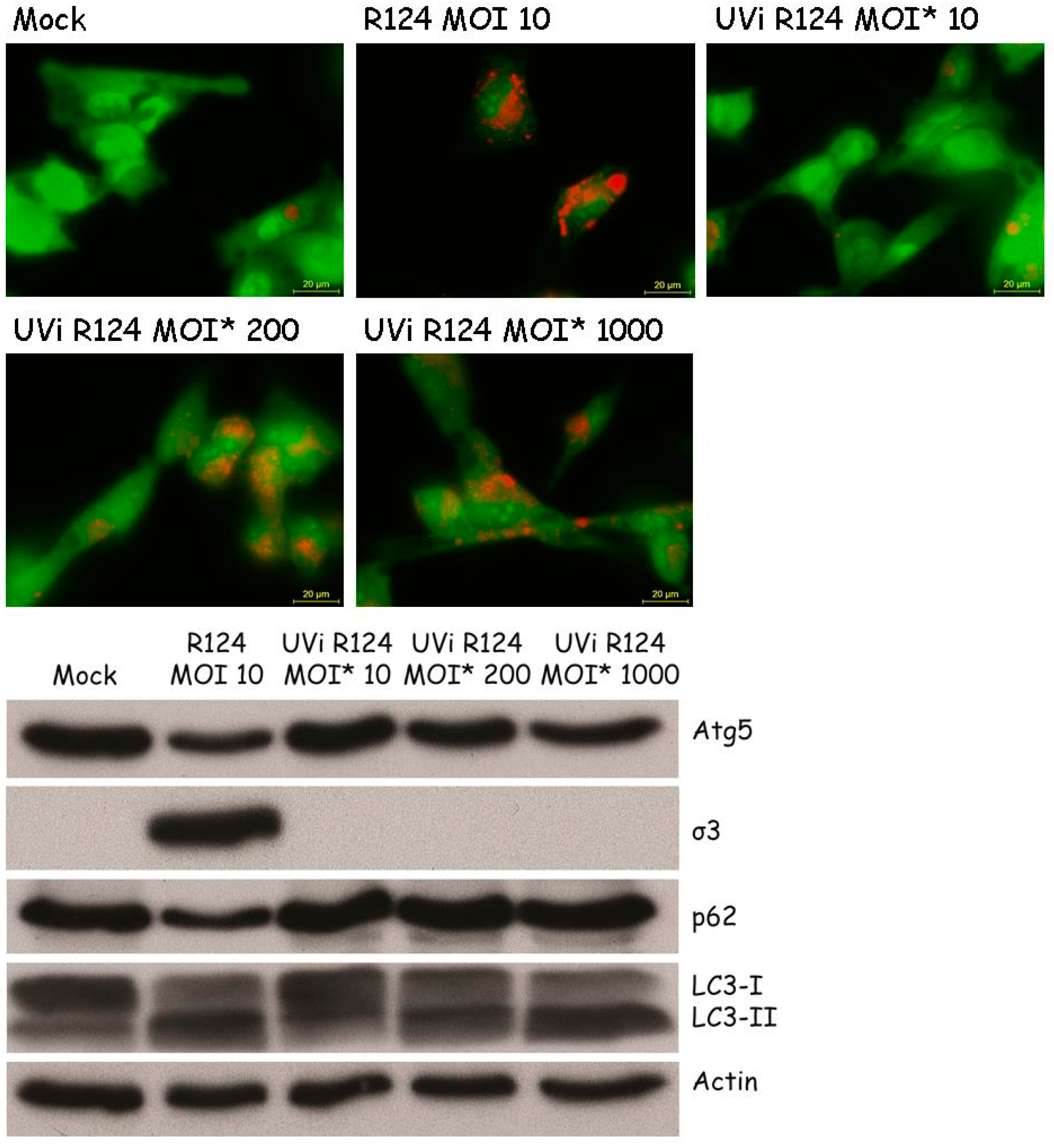
3.6. Reovirus Replication Facilitates the Induction of Acidic Vesicular Organelles, Atg5-Atg12 Conjugation, p62 Degradation and LC3 Lipidation in U87-MG Cells
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, X.; Chester, C.; Rajasekaran, N.; He, Z.; Kohrt, H.E. Strategic combinations: The future of oncolytic virotherapy with reovirus. Mol. Cancer Ther. 2016, 15, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Lennemann, N.J.; Coyne, C.B. Catch me if you can: The link between autophagy and viruses. PLoS Pathog. 2015, 11, e1004685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daussy, C.F.; Beaumelle, B.; Espert, L. Autophagy restricts HIV-1 infection. Oncotarget 2015, 6, 20752–20753. [Google Scholar] [PubMed]
- Campbell, G.R.; Spector, S.A. Inhibition of human immunodeficiency virus type-1 through autophagy. Curr. Opin. Microbiol. 2013, 16, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Levine, B. Autophagy and viruses: Adversaries or allies? J. Innate Immun. 2013, 5, 480–493. [Google Scholar] [PubMed]
- Jiang, H.; White, E.J.; Rios-Vicil, C.I.; Xu, J.; Gomez-Manzano, C.; Fueyo, J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J. Virol. 2011, 85, 4720–4729. [Google Scholar] [CrossRef] [PubMed]
- Arnoldi, F.; de Lorenzo, G.; Mano, M.; Schraner, E.M.; Wild, P.; Eichwald, C.; Burrone, O.R. Rotavirus increases levels of lipidated LC3 supporting accumulation of infectious progeny virus without inducing autophagosome formation. PLoS ONE 2014, 9, e95197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, P.I.; Huang, W.R.; Lai, I.H.; Cheng, C.Y.; Liu, H.J. The p17 nonstructural protein of avian reovirus triggers autophagy enhancing virus replication via activation of phosphatase and tensin deleted on chromosome 10 (PTEN) and AMP-activated protein kinase (AMPK), as well as dsRNA-dependent protein kinase (PKR)/eIF2α signaling pathways. J. Biol. Chem. 2013, 288, 3571–3584. [Google Scholar] [PubMed]
- Lv, S.; Xu, Q.; Sun, E.; Yang, T.; Li, J.; Feng, Y.; Zhang, Q.; Wang, H.; Zhang, J.; Wu, D. Autophagy activated by bluetongue virus infection plays a positive role in its replication. Viruses 2015, 7, 4657–4675. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus modulates autophagy during oncolysis of multiple myeloma. Autophagy 2013, 9, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin. Cancer Res. 2012, 18, 4962–4972. [Google Scholar] [CrossRef] [PubMed]
- Dubridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human-cells by using an epstein-barr-virus shuttle system. Mol. Cell Biol. 1987, 7, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Fallaux, F.J.; Kranenburg, O.; Cramer, S.J.; Houweling, A.; van Ormondt, H.; Hoeben, R.C.; van der Eb, A.J. Characterization of 911: A new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum. Gene Ther. 1996, 7, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Sou, Y.S.; Waguri, S.; Iwata, J.; Ueno, T.; Fujimura, T.; Hara, T.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Hieke, N.; Loffler, A.S.; Kaizuka, T.; Berleth, N.; Bohler, P.; Driessen, S.; Stuhldreier, F.; Friesen, O.; Assani, K.; Schmitz, K.; et al. Expression of a ULK1/2 binding-deficient ATG13 variant can partially restore autophagic activity in ATG13-deficient cells. Autophagy 2015, 11, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; van den Hengel, S.K.; Cramer, S.J.; de Groot, R.J.; Hoeben, R.C. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS ONE 2012, 7, e48064. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Mann, M.A.; Fields, B.N.; Tyler, K.L. Monoclonal-antibodies to reovirus reveal structure-function-relationships between capsid proteins and genetics of susceptibility to antibody action. J. Virol. 1991, 65, 6772–6781. [Google Scholar] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Stencel, J.E.; Liu, Y.; Blaum, B.S.; Reiter, D.M.; Feizi, T.; Dermody, T.S.; Stehle, T. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog. 2012, 8, e1003078. [Google Scholar] [CrossRef] [PubMed]
- Kemp, V.; Dautzenberg, I.J.; van den Wollenberg, D.J.; Hoeben, R.C. Autophagy has minor effects on reovirus-induced cytolysis. Unpublished data. 2017. [Google Scholar]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; Ros, W.; Lipinska, A.D.; van den Hengel, S.K.; Hoeben, R.C. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther. 2015, 22, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Xu, Q.Y.; Sun, E.C.; Zhang, J.K.; Wu, D.L. Dissection and integration of the autophagy signaling network initiated by bluetongue virus infection: Crucial candidates ERK1/2, Akt and AMPK. Sci. Rep. 2016, 6, 23130. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-β signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Dang, W.; Zhou, X.; Xu, L.; Wang, W.; Cao, W.; Chen, S.; Su, J.; Cai, X.; Xiao, S.; et al. PI3K-Akt-mTOR axis sustains rotavirus infection via the 4E-BP1 mediated autophagy pathway and represents an antiviral target. Virulence 2017. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Jackson, W.T. That which does not degrade you makes you stronger: Infectivity of poliovirus depends on vesicle acidification. Autophagy 2013, 9, 806–807. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Jackson, W.T. How positive-strand RNA viruses benefit from autophagosome maturation. J. Virol. 2013, 87, 9966–9972. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Duan, Y.; Han, C.; Yao, S.; Qi, X.; Gao, Y.; Maier, H.J.; Britton, P.; Chen, L.; Zhang, L.; et al. Infectious bursal disease virus subverts autophagic vacuoles to promote viral maturation and release. J. Virol. 2017, 91, e01883-16. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Stanifer, M.; Kural, C.; Cureton, D.K.; Massol, R.; Nibert, M.L.; Kirchhausen, T. Similar uptake but different trafficking and escape routes of reovirus virions and infectious subvirion particles imaged in polarized Madin-Darby canine kidney cells. Mol. Biol. Cell 2013, 24, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.G.; Guinea, R.; Benavente, J.; Carrasco, L. The entry of reovirus into L cells is dependent on vacuolar proton-ATPase activity. J. Virol. 1996, 70, 576–579. [Google Scholar] [PubMed]
- Bestebroer, J.; V’Kovski, P.; Mauthe, M.; Reggiori, F. Hidden behind autophagy: The unconventional roles of ATG proteins. Traffic 2013, 14, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Langereis, M.; Jung, J.; Zhou, X.; Jones, A.; Omta, W.; Tooze, S.A.; Stork, B.; Paludan, S.R.; Ahola, T.; et al. An siRNA screen for ATG protein depletion reveals the extent of the unconventional functions of the autophagy proteome in virus replication. J. Cell Biol. 2016, 214, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Reggiori, F. ATG proteins: Are we always looking at autophagy? Autophagy 2016, 12, 2502–2503. [Google Scholar] [CrossRef] [PubMed]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Moloughney, J.G.; Monken, C.E.; Tao, H.; Zhang, H.; Thomas, J.D.; Lattime, E.C.; Jin, S.V. Vaccinia virus leads to ATG12–ATG3 conjugation and deficiency in autophagosome formation. Autophagy 2014, 7, 1434–1447. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. ATG12-ATG3 connects basal autophagy and late endosome function. Autophagy 2015, 11, 961–962. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [PubMed]
- Murrow, L.; Malhotra, R.; Debnath, J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat. Cell Biol. 2015, 17, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Mainou, B.A.; Dermody, T.S. Transport to late endosomes is required for efficient reovirus infection. J. Virol. 2012, 86, 8346–8358. [Google Scholar] [CrossRef] [PubMed]
- Thete, D.; Danthi, P. Conformational changes required for reovirus cell entry are sensitive to pH. Virology 2015, 483, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Pan, L.Z.; Garant, K.; Pan, D.; Lee, P.W. Oncogenic Ras promotes reovirus spread by suppressing IFN-β production through negative regulation of RIG-I signaling. Cancer Res. 2010, 70, 4912–4921. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L.; Squier, M.K.; Rodgers, S.E.; Schneider, B.E.; Oberhaus, S.M.; Grdina, T.A.; Cohen, J.J.; Dermody, T.S. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J. Virol. 1995, 69, 6972–6979. [Google Scholar] [PubMed]
- Berger, A.K.; Hiller, B.E.; Thete, D.; Snyder, A.J.; Perez, E., Jr.; Upton, J.W.; Danthi, P. Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J. Virol. 2017, 91, e02404-16. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kemp, V.; Dautzenberg, I.J.C.; Limpens, R.W.; Van den Wollenberg, D.J.M.; Hoeben, R.C. Oncolytic Reovirus Infection Is Facilitated by the Autophagic Machinery. Viruses 2017, 9, 266. https://doi.org/10.3390/v9100266
Kemp V, Dautzenberg IJC, Limpens RW, Van den Wollenberg DJM, Hoeben RC. Oncolytic Reovirus Infection Is Facilitated by the Autophagic Machinery. Viruses. 2017; 9(10):266. https://doi.org/10.3390/v9100266
Chicago/Turabian StyleKemp, Vera, Iris J. C. Dautzenberg, Ronald W. Limpens, Diana J. M. Van den Wollenberg, and Rob C. Hoeben. 2017. "Oncolytic Reovirus Infection Is Facilitated by the Autophagic Machinery" Viruses 9, no. 10: 266. https://doi.org/10.3390/v9100266
APA StyleKemp, V., Dautzenberg, I. J. C., Limpens, R. W., Van den Wollenberg, D. J. M., & Hoeben, R. C. (2017). Oncolytic Reovirus Infection Is Facilitated by the Autophagic Machinery. Viruses, 9(10), 266. https://doi.org/10.3390/v9100266