Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
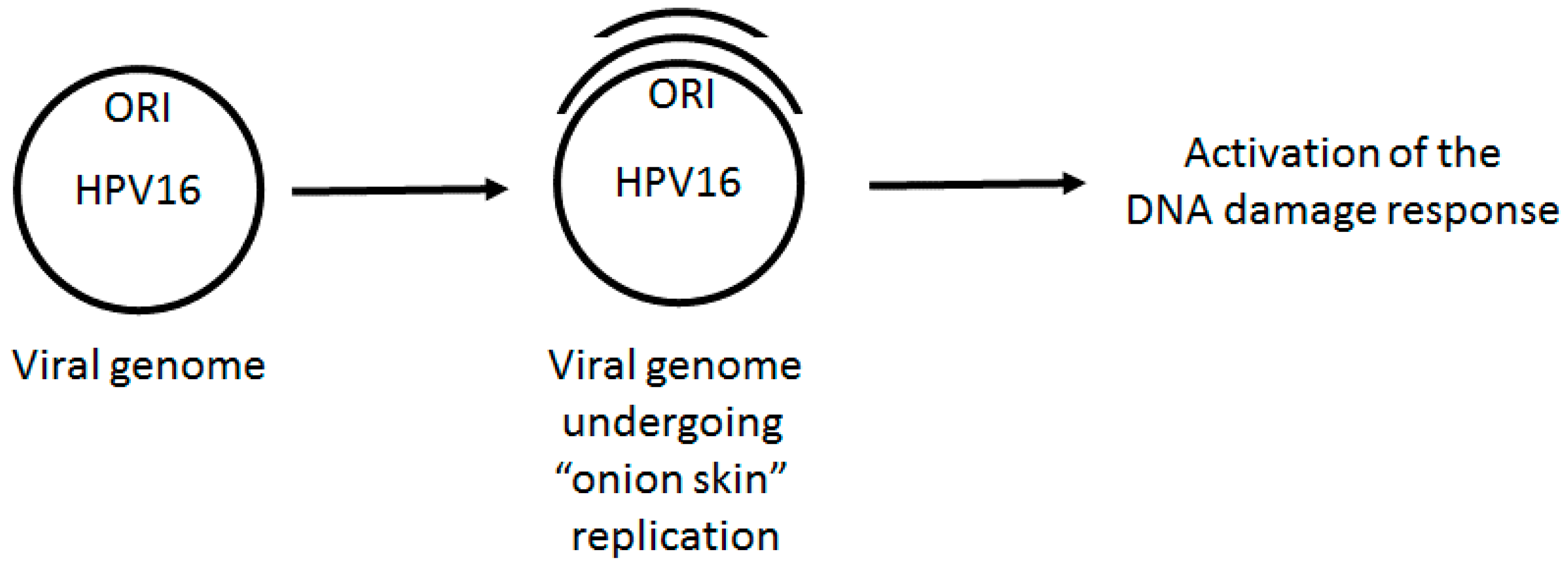
2. HPV DNA Replication
3. HPV Replication Activates the DDR
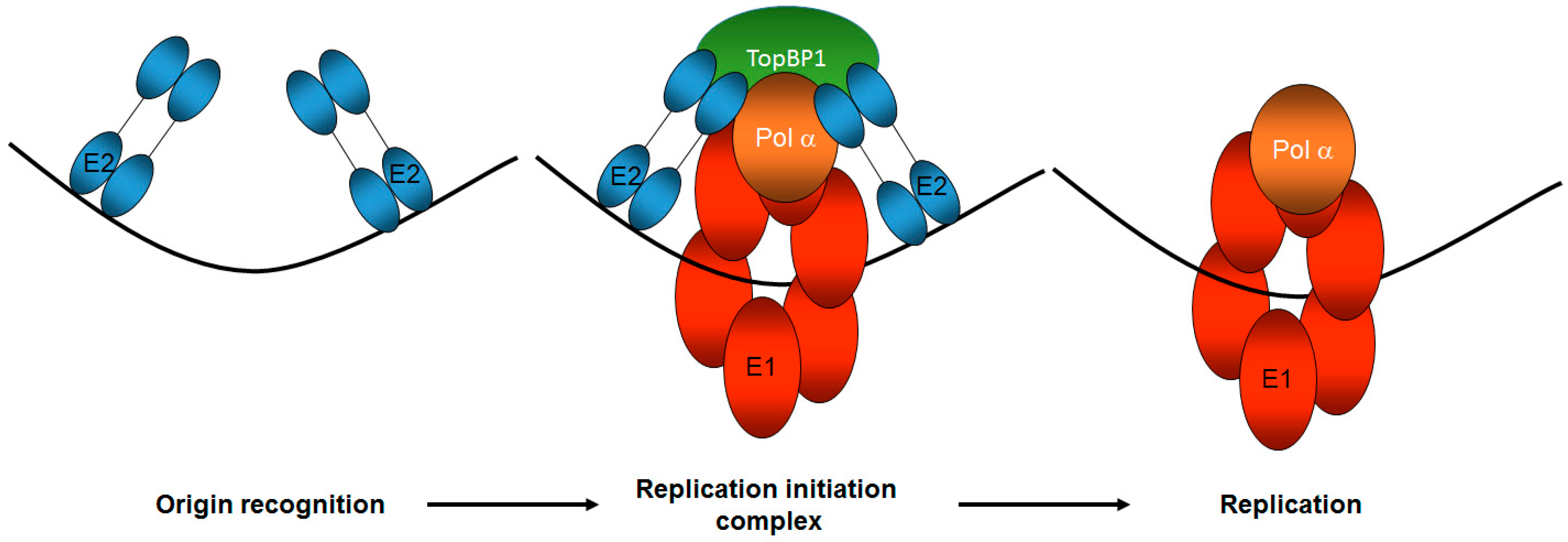
4. The Role of Viral Replication Proteins in Activating the DDR
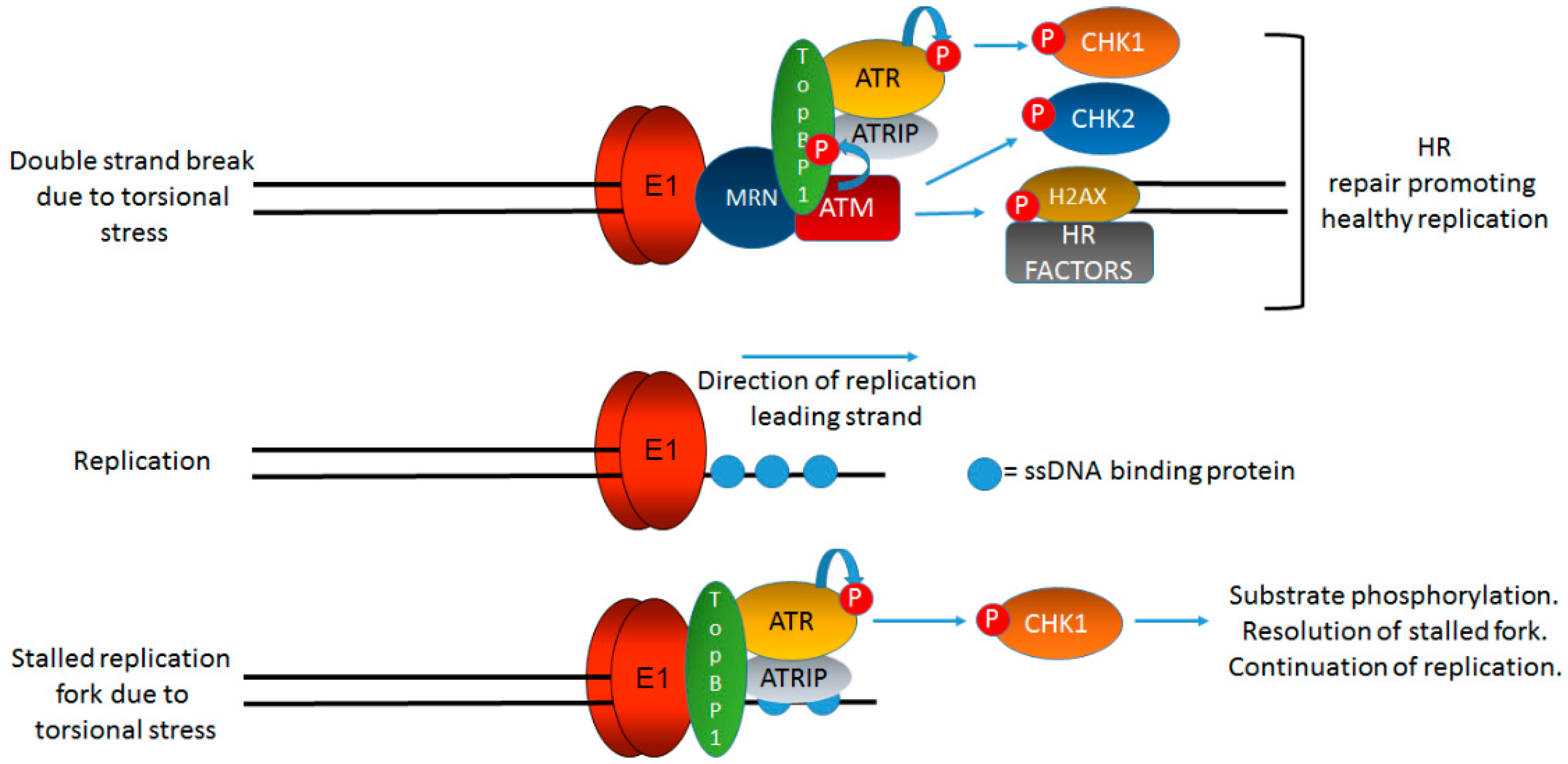
5. Formation of the E1–E2 Replication Complex Promotes the Activation of the ATM and ATR DDR Pathways: The Role of TopBP1
6. The Role of SIRT1 in the Viral Life Cycle
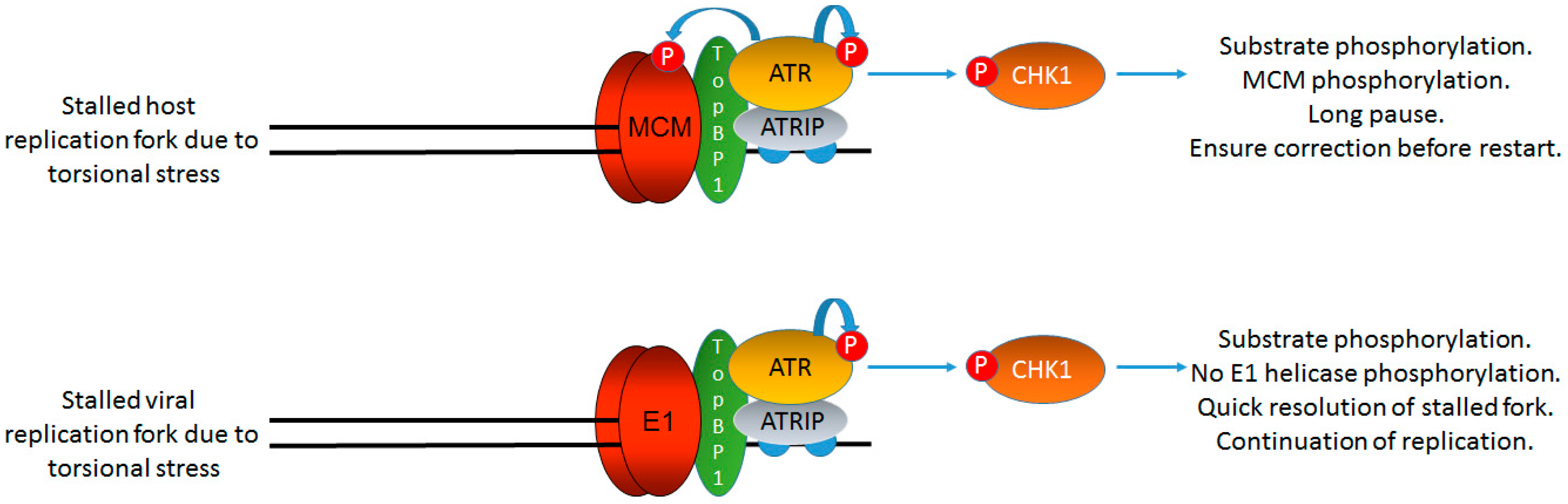
7. Why Do the DDR Signals Generated by HPV Not Arrest Viral Replication or Host Replication?
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L. Human papillomavirus-associated head and neck cancer is a distinct epidemiologic, clinical, and molecular entity. Semin. Oncol. 2004, 31, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Shah, K.V. Human papillomavirus-associated head and neck squamous cell carcinoma: mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Curr. Opin. Oncol. 2001, 13, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.M.; Laimins, L.A. Human papillomaviruses: Basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 2006, 16, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 2009, 384, 375–379. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- White, P.W.; Faucher, A.M.; Massariol, M.J.; Welchner, E.; Rancourt, J.; Cartier, M.; Archambault, J. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486. Antimicrob. Agents Chemother. 2005, 49, 4834–4842. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Coulombe, R.; Cameron, D.R.; Thauvette, L.; Massariol, M.J.; Amon, L.M.; Fink, D.; Titolo, S.; Welchner, E.; Yoakim, C.; et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J. Biol. Chem. 2004, 279, 6976–6985. [Google Scholar] [CrossRef] [PubMed]
- Archambault, J.; Melendy, T. Targeting human papillomavirus genome replication for antiviral drug discovery. Antivir. Ther. 2013, 18, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Cullen, A.P.; Reid, R.; Campion, M.; Lorincz, A.T. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J. Virol. 1991, 65, 606–612. [Google Scholar] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Cooper, K.; Herrington, C.S.; Lo, E.S.; Evans, M.F.; McGee, J.O. Integration of human papillomavirus types 16 and 18 in cervical adenocarcinoma. J. Clin. Pathol. 1992, 45, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [PubMed]
- Durst, M.; Croce, C.M.; Gissmann, L.; Schwarz, E.; Huebner, K. Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas. Proc. Natl. Acad. Sci. USA 1987, 84, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Ramqvist, T.; Mints, M.; Tertipis, N.; Nasman, A.; Romanitan, M.; Dalianis, T. Studies on human papillomavirus (HPV) 16 E2, E5 and E7 mRNA in HPV-positive tonsillar and base of tongue cancer in relation to clinical outcome and immunological parameters. Oral Oncol. 2015, 51, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Nulton, T.J.; Olex, A.L.; Dozmorov, M.; Morgan, I.M.; Windle, B. Analysis of the cancer genome atlas sequencing data reveals novel properties of the human papillomavirus 16 genome in head and neck squamous cell carcinoma. Oncotarget 2017, 8, 17684–17699. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Kowalczyk, A.; Chen, H.; Roeder, G.E.; Sessions, R.; Buckle, M.; Gaston, K. E2 proteins from high- and low-risk human papillomavirus types differ in their ability to bind p53 and induce apoptotic cell death. J. Virol. 2006, 80, 4580–4590. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Naeger, L.K.; DiMaio, D. Activation of the endogenous p53 growth inhibitory pathway in HeLa cervical carcinoma cells by expression of the bovine papillomavirus E2 gene. Oncogene 1996, 12, 795–803. [Google Scholar] [PubMed]
- Naeger, L.K.; Goodwin, E.C.; Hwang, E.S.; DeFilippis, R.A.; Zhang, H.; DiMaio, D. Bovine papillomavirus E2 protein activates a complex growth-inhibitory program in p53-negative HT-3 cervical carcinoma cells that includes repression of cyclin A and cdc25A phosphatase genes and accumulation of hypophosphorylated retinoblastoma protein. Cell Growth Differ. 1999, 10, 413–422. [Google Scholar] [PubMed]
- Johung, K.; Goodwin, E.C.; DiMaio, D. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J. Virol. 2007, 81, 2102–2116. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; DiMaio, D. Induced senescence in HeLa cervical carcinoma cells containing elevated telomerase activity and extended telomeres. Cell Growth Differ. 2001, 12, 525–534. [Google Scholar] [PubMed]
- Hwang, E.S.; Riese, D.J., 2nd; Settleman, J.; Nilson, L.A.; Honig, J.; Flynn, S.; DiMaio, D. Inhibition of cervical carcinoma cell line proliferation by the introduction of a bovine papillomavirus regulatory gene. J. Virol. 1993, 67, 3720–3729. [Google Scholar] [PubMed]
- Magaldi, T.G.; Almstead, L.L.; Bellone, S.; Prevatt, E.G.; Santin, A.D.; DiMaio, D. Primary human cervical carcinoma cells require human papillomavirus E6 and E7 expression for ongoing proliferation. Virology 2012, 422, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; Yang, E.; Lee, C.J.; Lee, H.W.; DiMaio, D.; Hwang, E.S. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 10978–10983. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; DiMaio, D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 12513–12518. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; Naeger, L.K.; Breiding, D.E.; Androphy, E.J.; DiMaio, D. Transactivation-competent bovine papillomavirus E2 protein is specifically required for efficient repression of human papillomavirus oncogene expression and for acute growth inhibition of cervical carcinoma cell lines. J. Virol. 1998, 72, 3925–3934. [Google Scholar] [PubMed]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic instability of the host cell induced by the human papillomavirus replication machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Gautam, D.; Ok, S.T.; Johnson, B.A.; Anacker, D.C.; Moody, C.A. Homologous recombination repair factors, Rad51 and BRCA1, are necessary for productive replication of human papillomavirus 31. J. Virol. 2015, 90, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Kim, B.; Moody, C.A. HPV31 utilizes the ATR-Chk1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Moody, C.A.; Weitzman, M.D.; Weitzman, J.B.; Moody, C.A.; Laimins, L.A.; Hong, S.; Dutta, A.; Laimins, L.A.; Gillespie, K.A.; et al. Impact of the DNA damage response on human papillomavirus chromatin. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017, 231, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Aloor, H.L.; Moody, C.A. The Rb binding domain of HPV31 E7 is required to maintain high levels of DNA repair factors in infected cells. Virology 2017, 500, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Bristol, M.L.; Wang, X.; Smith, N.W.; Son, M.P.; Evans, M.R.; Morgan, I.M. DNA damage reduces the quality, but not the quantity of human papillomavirus 16 E1 and E2 DNA replication. Viruses 2016, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 regulates transcription of the topoisomerase IIβ-binding protein 1 (TopBP1) gene to activate the ATR pathway and promote human papillomavirus replication. MBio 2015, 6, e02006-15. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Laimins, L.A. Human papillomavirus and the DNA damage response: Exploiting host repair pathways for viral replication. Viruses 2017, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Melendy, T.; Sedman, J.; Stenlund, A. Cellular factors required for papillomavirus DNA replication. J. Virol. 1995, 69, 7857–7867. [Google Scholar] [PubMed]
- Hu, Y.; Clower, R.V.; Melendy, T. Cellular topoisomerase I modulates origin binding by bovine papillomavirus type 1 E1. J. Virol. 2006, 80, 4363–4371. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Loo, Y.M.; Militello, K.T.; Melendy, T. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A. J. Virol. 1999, 73, 4899–4907. [Google Scholar] [PubMed]
- Clower, R.V.; Fisk, J.C.; Melendy, T. Papillomavirus E1 protein binds to and stimulates human topoisomerase I. J. Virol. 2006, 80, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Melendy, T. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J. Virol. 2004, 78, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Masterson, P.J.; Stanley, M.A.; Lewis, A.P.; Romanos, M.A. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase α-primase p68 subunit. J. Virol. 1998, 72, 7407–7419. [Google Scholar] [PubMed]
- Baran, N.; Neer, A.; Manor, H. “Onion Skin” replication of integrated polyoma virus DNA and flanking sequences in polyoma-transformed rat cells: Termination within a specific cellular DNA segment. Proc. Natl. Acad. Sci. USA 1983, 80, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Gerspach, R.; Matz, B. Herpes simplex virus-induced “Rolling Circle” amplification of SV40 DNA sequences in a transformed hamster cell line correlates with tandem integration of the SV40 genome. Virology 1989, 173, 723–727. [Google Scholar] [CrossRef]
- Mannik, A.; Runkorg, K.; Jaanson, N.; Ustav, M.; Ustav, E. Induction of the bovine papillomavirus origin “Onion Skin”-type DNA replication at high E1 protein concentrations in vivo. J. Virol. 2002, 76, 5835–5845. [Google Scholar] [CrossRef] [PubMed]
- Syu, L.J.; Fluck, M.M. Site-specific in situ amplification of the integrated polyomavirus genome: A case for a context-specific over-replication model of gene amplification. J. Mol. Biol. 1997, 271, 76–99. [Google Scholar] [CrossRef] [PubMed]
- Stary, A.; Sarasin, A. Simian virus 40 (SV40) large T antigen-dependent amplification of an epstein-barr virus-SV40 hybrid shuttle vector integrated into the human HeLa cell genome. J. Gen. Virol. 1992, 73, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Komeda, Y.; Umemori, T.; Kubota, Y.; Takisawa, H.; Araki, H. Efficient initiation of DNA replication in eukaryotes requires Dpb11/TopBP1-GINS interaction. Mol. Cell. Biol. 2013, 33, 2614–2622. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 2010, 140, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Direct regulation of treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 2011, 193, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 2007, 445, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Umemori, T.; Hirai, K.; Muramatsu, S.; Kamimura, Y.; Araki, H. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 2007, 445, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Takisawa, H. Xenopus Cut5 is essential for a CDK-dependent process in the initiation of DNA replication. EMBO J. 2003, 22, 2526–2535. [Google Scholar] [CrossRef] [PubMed]
- Balestrini, A.; Cosentino, C.; Errico, A.; Garner, E.; Costanzo, V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat. Cell Biol. 2010, 12, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Mackintosh, L.J.; Bodily, J.M.; Dornan, E.S.; Laimins, L.A.; Morgan, I.M. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J. Virol. 2012, 86, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2 mediated DNA replication. J. Virol. 2015, 89, 17684–17699. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.; Glover, J.N. BRCT domains: Easy as one, two, three. Cell Cycle 2011, 10, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, C.P.; Carr, A.M.; Oliver, A.W. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst.) 2014, 22, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Duursma, A.M.; Driscoll, R.; Elias, J.E.; Cimprich, K.A. A role for the MRN complex in ATR activation via TOPBP1 recruitment. Mol. Cell 2013, 50, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Cortez, D. ATR signaling: More than meeting at the fork edward. Biochem. J. 2013, 436, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Shigechi, T.; Tomida, J.; Sato, K.; Kobayashi, M.; Eykelenboom, J.K.; Pessina, F.; Zhang, Y.; Uchida, E.; Ishiai, M.; Lowndes, N.F.; et al. ATR-ATRIP kinase complex triggers activation of the fanconi anemia DNA repair pathway. Cancer Res. 2012, 72, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Leffak, M. ATRIP from TopBP1 to ATR-in vitro activation of a DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 2010, 107, 13561–13562. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Takeishi, Y.; Ueda, S.; Tsurimoto, T. Interaction between Rad9-Hus1-Rad1 and TopBP1 activates ATR-ATRIP and promotes TopBP1 recruitment to sites of UV-damage. DNA Repair (Amst.) 2014, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wardlaw, C.P.; Morishita, T.; Miyabe, I.; Chahwan, C.; Caspari, T.; Schmidt, U.; Carr, A.M.; Garcia, V. The Rad4(TopBP1) ATR-activation domain functions in G1/S phase in a chromatin-dependent manner. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Ataxia-telangiectasia mutated (ATM)-dependent activation of ATR occurs through phosphorylation of TopBP1 by ATM. J. Biol. Chem. 2007, 282, 17501–17506. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Mochan, T.A.; Venere, M.; DiTullio, R.A., Jr.; Halazonetis, T.D. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003, 63, 8586–8591. [Google Scholar] [PubMed]
- Yoo, H.Y.; Kumagai, A.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. The Mre11-Rad50-Nbs1 complex mediates activation of TopBP1 by ATM. Mol. Biol. Cell 2009, 20, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, C.; Li, T.; Bruhn, C.; Krueger, A.; Min, W.; Wang, Z.; Carr, A.M. An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lin, F.T.; Ruppert, J.M.; Lin, W.C. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol. Cell. Biol. 2003, 23, 3287–3304. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ling, S.; Lin, W. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [PubMed]
- Kanginakudru, S.; DeSmet, M.; Thomas, Y.; Morgan, I.M.; Androphy, E.J. Levels of the E2 interacting protein TopBP1 modulate papillomavirus maintenance stage replication. Virology 2015, 478, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorospe, M.; de Cabo, R. AsSIRTing the DNA damage response. Trends Cell Biol. 2008, 18, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lin, Y.; Leng, W.; Jung, S.Y.; Zhang, H.; Deng, M.; Evans, D.; Li, Y.; Luo, K.; Qin, B.; et al. A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Mol. Cell 2014, 56, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lahusen, T.J.; Chen, Q.; Xu, X.; Jenkins, L.M.M.; Leo, E.; Fu, H.; Aladjem, M.; Pommier, Y.; Appella, E.; et al. SIRT1 deacetylates TopBP1 and modulates intra-S-phase checkpoint and DNA replication origin firing. Int. J. Biol. Sci. 2014, 10, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Smith, N.; Wang, X.; Morgan, I.M. The deacetylase SIRT1 regulates the replication properties of human papillomavirus 16 E1 and E2. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Langsfeld, E.S.; Bodily, J.M.; Laimins, L.A. The deacetylase Sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and Rad51 to viral genomes. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhang, X.; Sengupta, N.; Lane, W.S.; Seto, E. SIRT1 regulates the function of the nijmegen breakage syndrome protein. Mol. Cell 2007, 27, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Csernok, A.; Aydin, S.; Kreienberg, R.; Wiesmuller, L.; Gatz, S.A. Role of SIRT1 in homologous recombination. DNA Repair (Amst.) 2010, 9, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 oncogenes: Key factors for viral carcinogenesis and therapeutic targets. Trends Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Fisk, J.C.; Dornan, E.S.; Donaldson, M.M.; Melendy, T.; Morgan, I.M. Human papillomavirus E1 and E2 mediated DNA replication is not arrested by DNA damage signalling. Virology 2010, 406, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Glick, G.; Elledge, S.J. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. USA 2004, 101, 10078–10083. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High risk α papillomavirus oncogenes impair the homologous recombination pathway. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Kwon, D.; McBride, A.A. Papillomavirus E2 proteins and the host BRD4 protein associate with transcriptionally active cellular chromatin. J. Virol. 2009, 83, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Brake, T.; Lambert, P.F. Estrogen contributes to the onset, persistence, and malignant progression of cervical cancer in a human papillomavirus-transgenic mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 2490–2495. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Shin, M.K.; Korach, K.S.; Lambert, P.F. Requirement for stromal estrogen receptor α in cervical neoplasia. Horm. Cancer 2013, 4, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Wiedmeyer, K.; Shai, A.; Korach, K.S.; Lambert, P.F. Requirement for estrogen receptor α in a mouse model for human papillomavirus-associated cervical cancer. Cancer Res. 2008, 68, 9928–9934. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Park, J.W.; Lambert, P.F.; Chung, S.H. Requirement of estrogen receptor α DNA-binding domain for HPV oncogene-induced cervical carcinogenesis in mice. Carcinogenesis 2014, 35, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Caldon, C.E. Estrogen signaling and the DNA damage response in hormone dependent breast cancers. Front. Oncol. 2014, 4, 106. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Liehr, J.G. Estrogen, DNA damage and mutations. Mutat. Res. 1999, 424, 107–115. [Google Scholar] [CrossRef]
- Liehr, J.G. Genotoxic effects of estrogens. Mutat. Res. 1990, 238, 269–276. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Evinger, A.J.; Lee, E.; Levin, E.R. Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair. Mol. Biol. Cell 2009, 20, 3374–3389. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar] [PubMed]
- Day, P.M.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J. Virol. 1998, 72, 142–150. [Google Scholar] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bristol, M.L.; Das, D.; Morgan, I.M. Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling. Viruses 2017, 9, 268. https://doi.org/10.3390/v9100268
Bristol ML, Das D, Morgan IM. Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling. Viruses. 2017; 9(10):268. https://doi.org/10.3390/v9100268
Chicago/Turabian StyleBristol, Molly L., Dipon Das, and Iain M. Morgan. 2017. "Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling" Viruses 9, no. 10: 268. https://doi.org/10.3390/v9100268
APA StyleBristol, M. L., Das, D., & Morgan, I. M. (2017). Why Human Papillomaviruses Activate the DNA Damage Response (DDR) and How Cellular and Viral Replication Persists in the Presence of DDR Signaling. Viruses, 9(10), 268. https://doi.org/10.3390/v9100268