
Antibody Competition Reveals Surface Location of HPV L2 Minor Capsid Protein Residues 17–36



Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of L2 mAbs
2.2. PsV/QV Particle Production and Purification
2.3. Production and Isolation of Native HPV in Organotypic Raft Culture
2.4. Titration of Native HPV by qPCR
2.5. Sequencing of L2 mAb Variable Regions
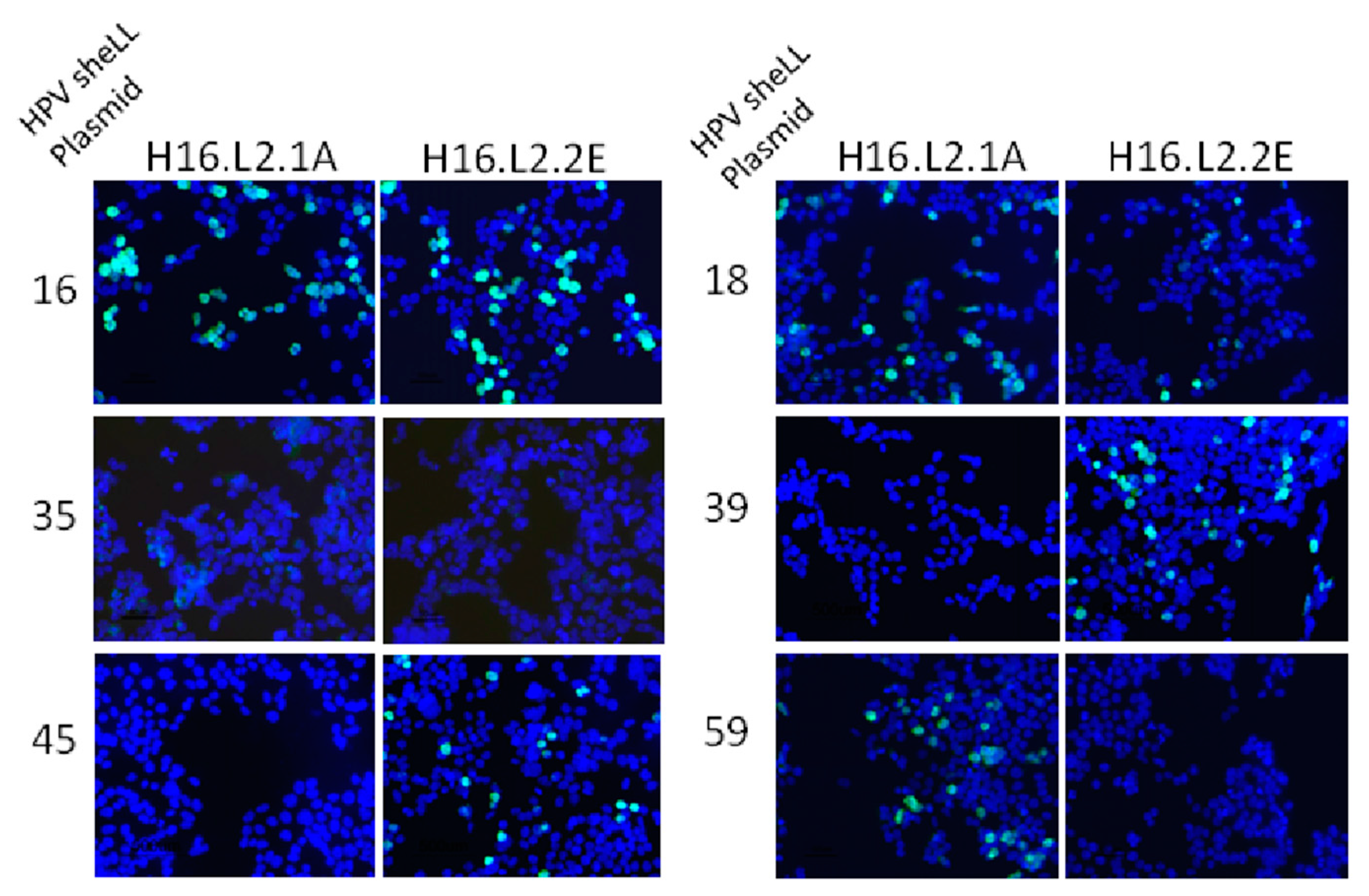
2.6. Immunofluorescence Assay
2.7. Direct Binding ELISAs
2.8. Peptide Binding ELISAs
2.9. Capture ELISAs
2.10. QV Post-Attachment Neutralization
2.11. Native HPV Neutralization Assay
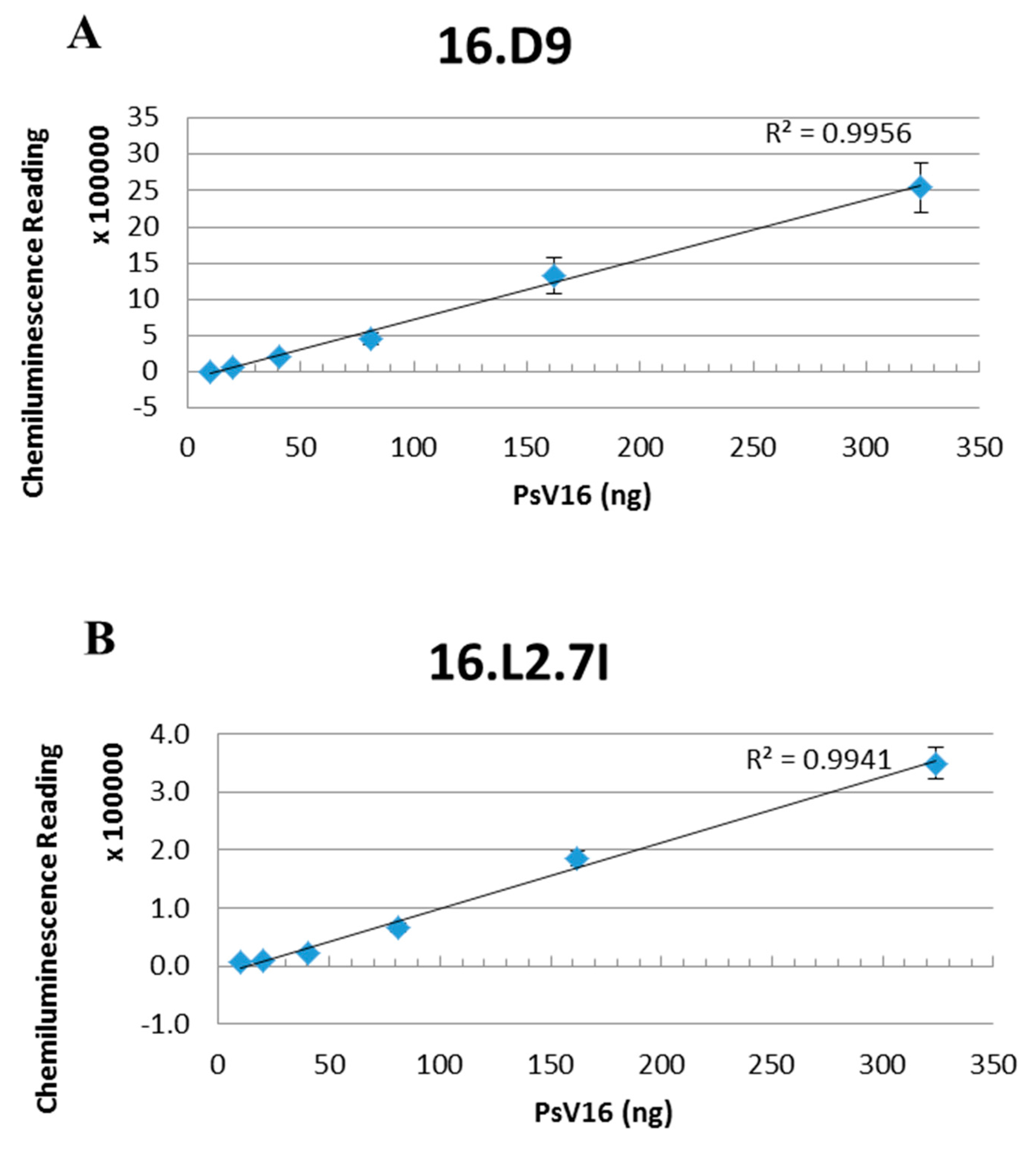
2.12. Quantitation of L2 Protein
3. Results
3.1. Sequencing Confirms L2 mAbs Are Separate Clones
3.2. L2 mAbs Demonstrate Cross-Reactive Binding
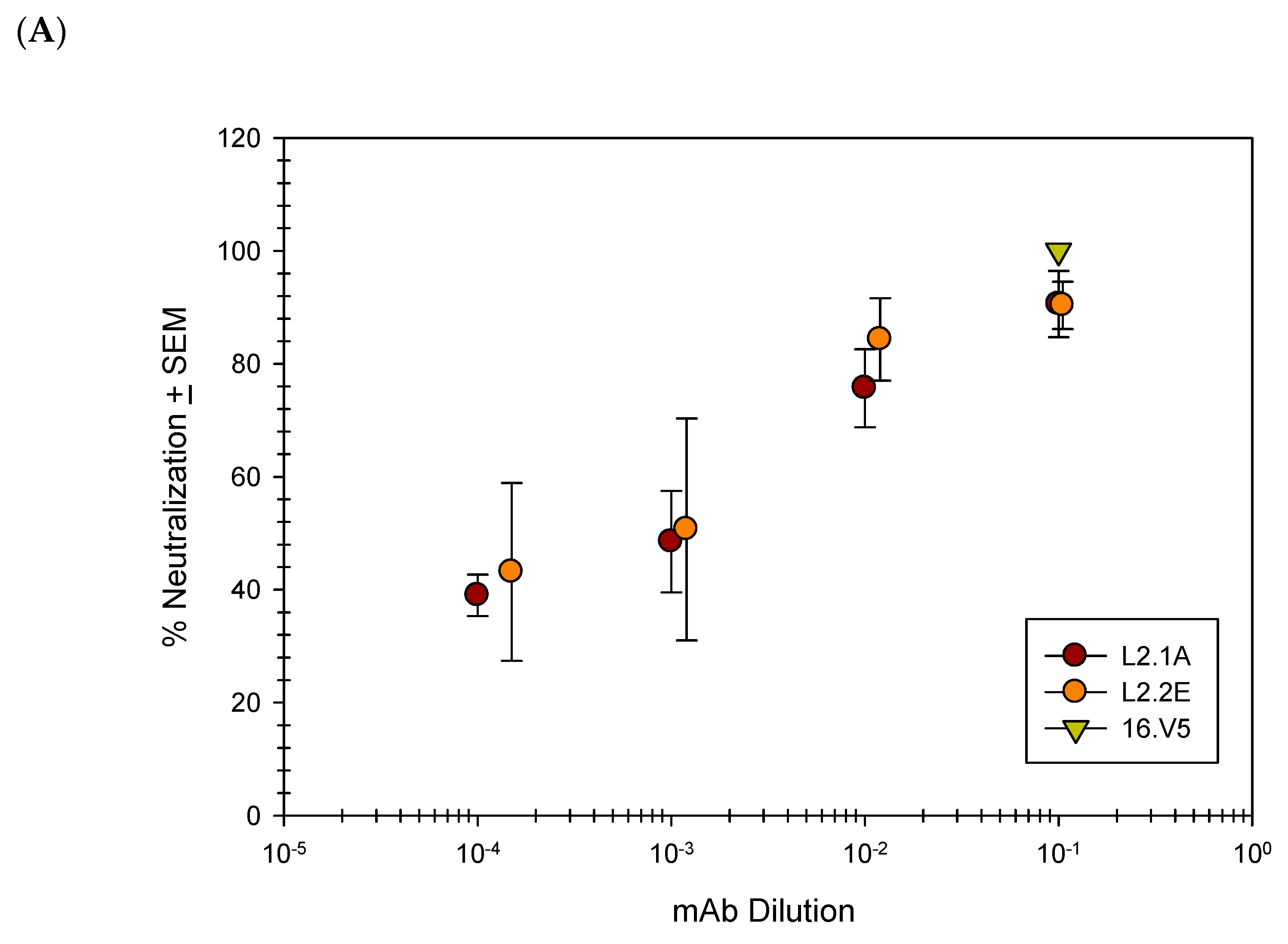
3.3. L2 mAbs Are Capable of Neutralizing Infection
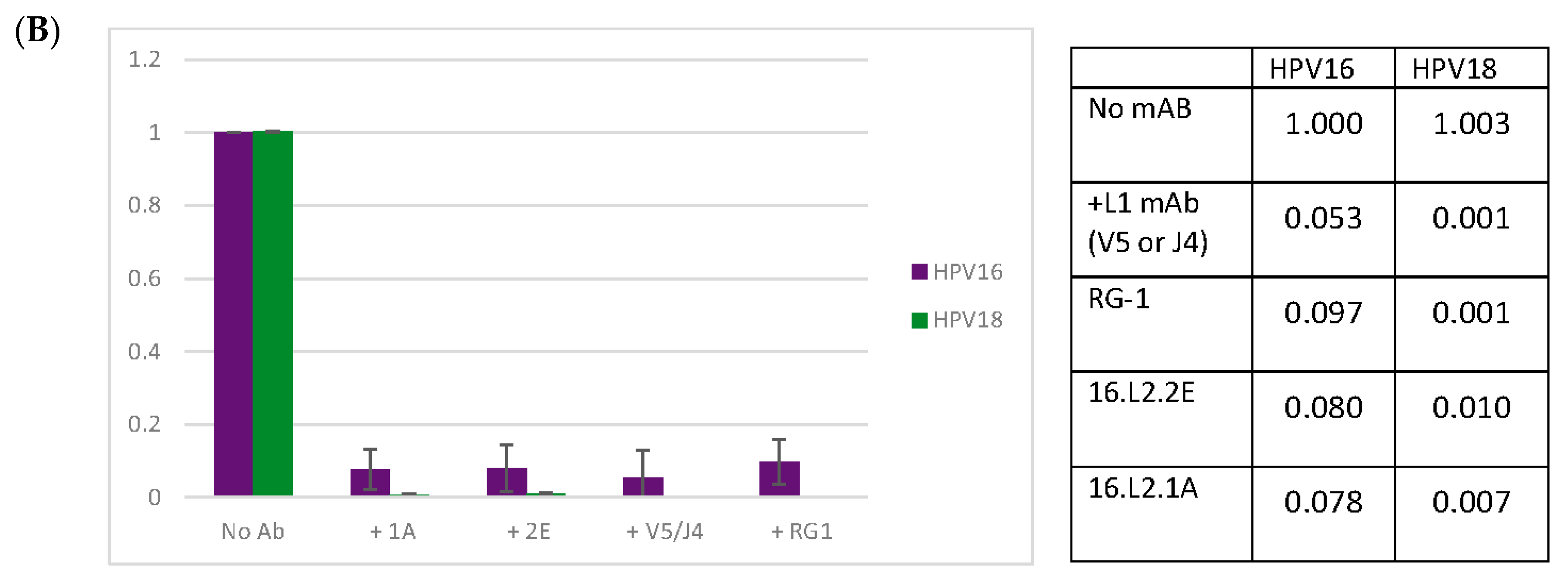
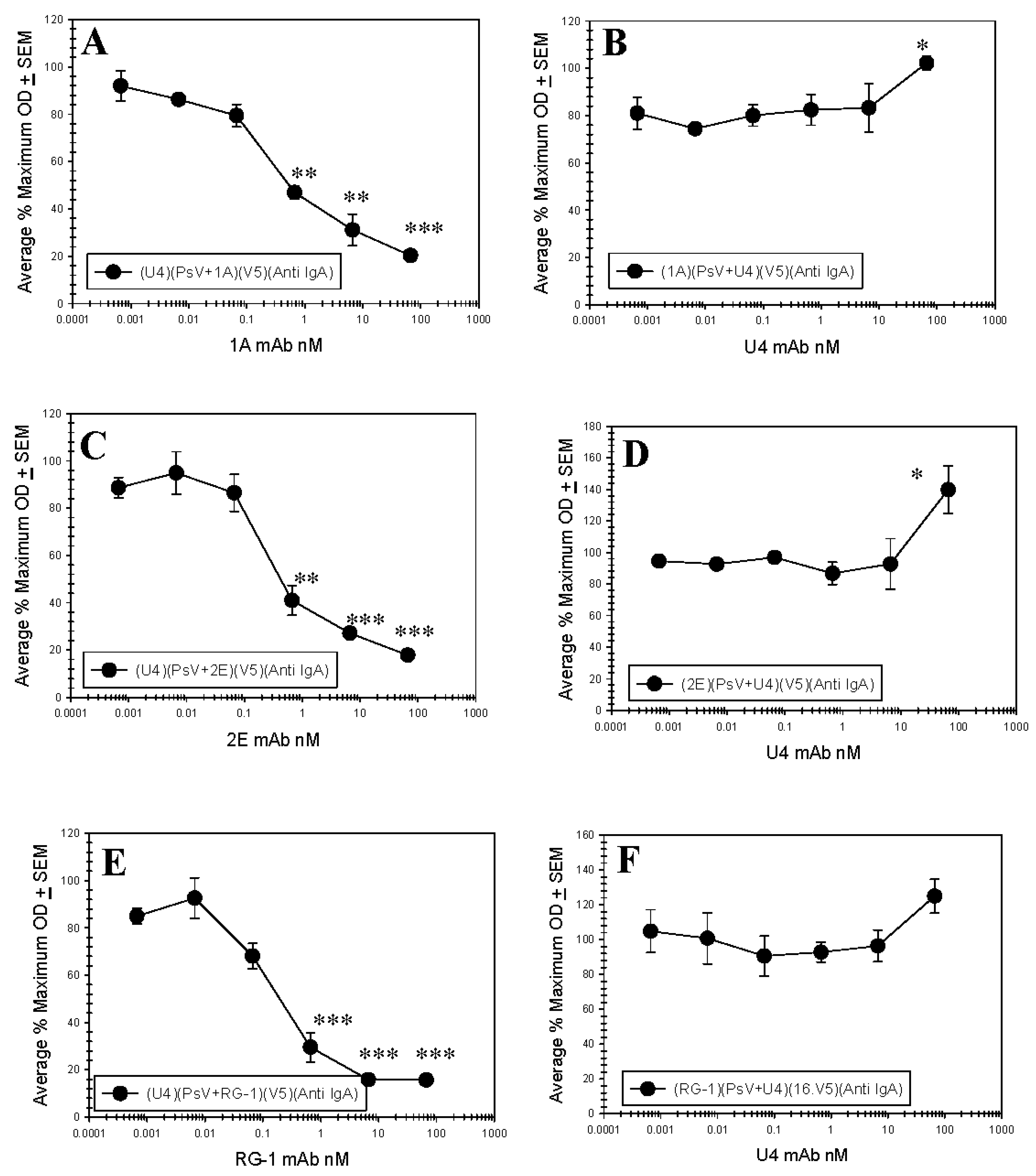
3.4. Anti-L1 Capture ELISAs Reveal U4 and Anti-L2 mAb Competition
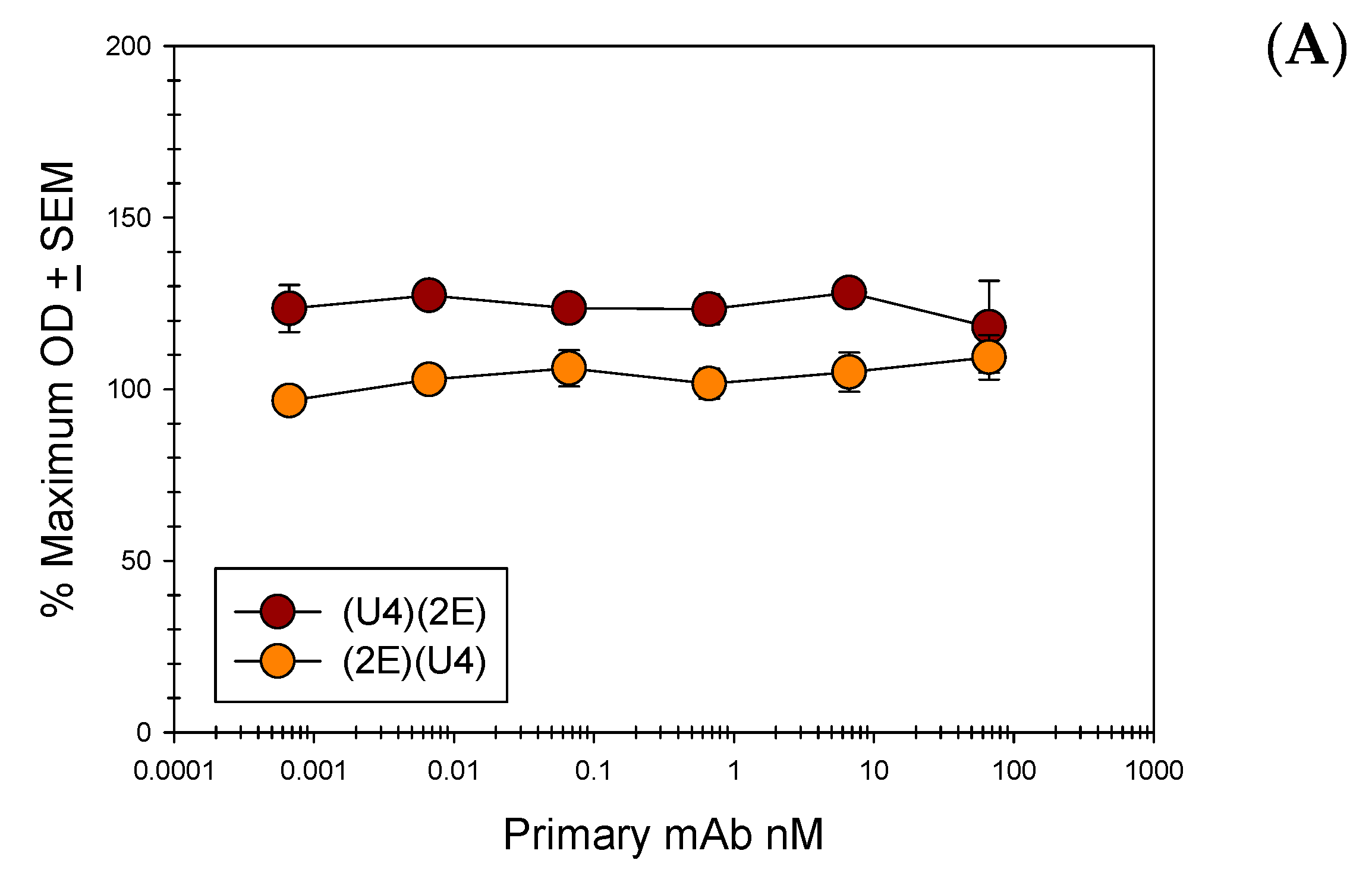
3.5. Anti-L2 Capture ELISAs Reveal the Accessibility of the L2 Epitope in the Presence of H16.U4
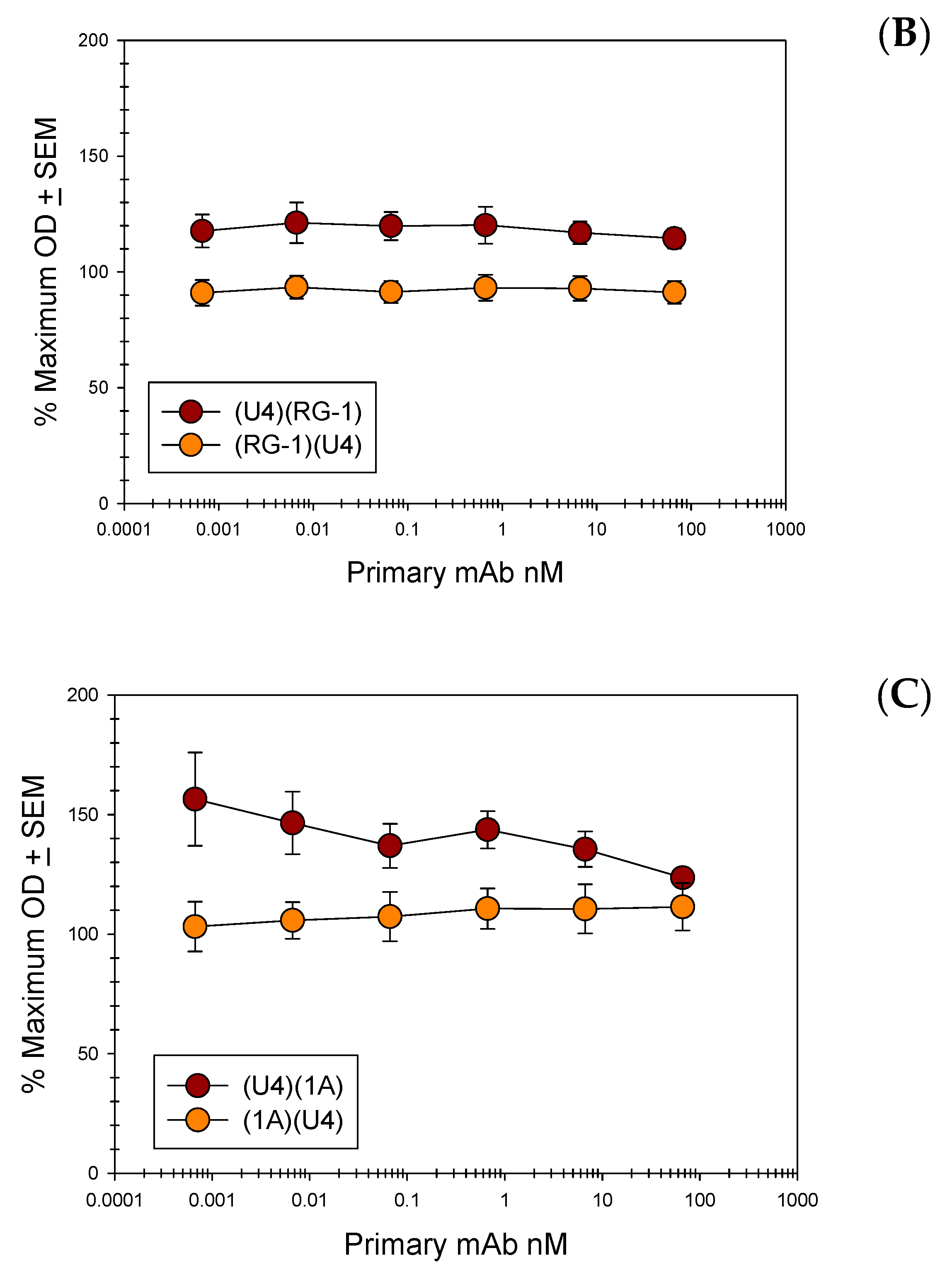
3.6. Direct ELISAs Alter Epitope Availability
3.7. L2 Quantification Agrees with Degree of U4 and 2E Competition
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
Gel Filtration Chromatography and Immunoblot
References
- De Villiers, E.M. Cross-roads in the classification of papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. 1), 2–23. [Google Scholar] [CrossRef] [PubMed]
- Garland, S.M.; Kjaer, S.K.; Munoz, N.; Block, S.L.; Brown, D.R.; DiNubile, M.J.; Lindsay, B.R.; Kuter, B.J.; Perez, G.; Dominiak-Felden, G.; et al. Impact and Effectiveness of the Quadrivalent Human Papillomavirus Vaccine: A Systematic Review of 10 Years of Real-world Experience. Clin. Infect. Dis. 2016, 63, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell 2000, 5, 557–567. [Google Scholar] [CrossRef]
- Christensen, N.D.; Kreider, J.W. Antibody-mediated neutralization in vivo of infectious papillomaviruses. J. Virol. 1990, 64, 3151–3157. [Google Scholar] [PubMed]
- Rose, R.C.; Bonnez, W.; Da Rin, C.; McCance, D.J.; Reichman, R.C. Serological differentiation of human papillomavirus types 11, 16 and 18 using recombinant virus-like particles. J. Gen. Virol. 1994, 75 Pt 9, 2445–2449. [Google Scholar] [CrossRef] [PubMed]
- Hines, J.F.; Ghim, S.J.; Christensen, N.D.; Kreider, J.W.; Barnes, W.A.; Schlegel, R.; Jenson, A.B. The expressed L1 proteins of HPV-1, HPV-6, and HPV-11 display type-specific epitopes with native conformation and reactivity with neutralizing and nonneutralizing antibodies. Pathobiology 1994, 62, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Hubbert, N.L.; Kirnbauer, R.; Christensen, N.D.; Lowy, D.R.; Schiller, J.T. Assessment of the serological relatedness of genital human papillomaviruses by hemagglutination inhibition. J. Virol. 1996, 70, 3298–3301. [Google Scholar] [PubMed]
- Kawana, K.; Matsumoto, K.; Yoshikawa, H.; Taketani, Y.; Kawana, T.; Yoshiike, K.; Kanda, T. A surface immunodeterminant of human papillomavirus type 16 minor capsid protein L2. Virology 1998, 245, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kawana, K.; Yoshikawa, H.; Taketani, Y.; Yoshiike, K.; Kanda, T. Common neutralization epitope in minor capsid protein L2 of human papillomavirus types 16 and 6. J. Virol. 1999, 73, 6188–6190. [Google Scholar] [PubMed]
- Gambhira, R.; Karanam, B.; Jagu, S.; Roberts, J.N.; Buck, C.B.; Bossis, I.; Alphs, H.; Culp, T.; Christensen, N.D.; Roden, R.B. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J. Virol. 2007, 81, 13927–13931. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.S.; O’Neil, B.W.; Grindlay, G.J.; Curtis, F.; Knowles, G.; Chandrachud, L. A peptide encoding a B-cell epitope from the N-terminus of the capsid protein L2 of bovine papillomavirus-4 prevents disease. Virology 1997, 234, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Chandrachud, L.M.; Grindlay, G.J.; McGarvie, G.M.; O’Neil, B.W.; Wagner, E.R.; Jarrett, W.F.; Campo, M.S. Vaccination of cattle with the N-terminus of L2 is necessary and sufficient for preventing infection by bovine papillomavirus-4. Virology 1995, 211, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Gaukroger, J.M.; Chandrachud, L.M.; O’Neil, B.W.; Grindlay, G.J.; Knowles, G.; Campo, M.S. Vaccination of cattle with bovine papillomavirus type 4 L2 elicits the production of virus-neutralizing antibodies. J. Gen. Virol. 1996, 77 Pt 7, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Weissinger, E.M.; Henderson, D.W.; Booy, F.; Kirnbauer, R.; Mushinski, J.F.; Lowy, D.R.; Schiller, J.T. Neutralization of bovine papillomavirus by antibodies to L1 and L2 capsid proteins. J. Virol. 1994, 68, 7570–7574. [Google Scholar] [PubMed]
- Christensen, N.D.; Kreider, J.W.; Kan, N.C.; DiAngelo, S.L. The open reading frame L2 of cottontail rabbit papillomavirus contains antibody-inducing neutralizing epitopes. Virology 1991, 181, 572–579. [Google Scholar] [CrossRef]
- Embers, M.E.; Budgeon, L.R.; Pickel, M.; Christensen, N.D. Protective immunity to rabbit oral and cutaneous papillomaviruses by immunization with short peptides of L2, the minor capsid protein. J. Virol. 2002, 76, 9798–9805. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Borenstein, L.A.; Selvakumar, R.; Ahmed, R.; Wettstein, F.O. Effective vaccination against papilloma development by immunization with L1 or L2 structural protein of cottontail rabbit papillomavirus. Virology 1992, 187, 612–619. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Gambhira, R.; Buck, C.B.; Pang, Y.Y.; Thompson, C.D.; Culp, T.D.; Christensen, N.D.; Lowy, D.R.; Schiller, J.T.; Roden, R.B. Cross-neutralization of cutaneous and mucosal Papillomavirus types with anti-sera to the amino terminus of L2. Virology 2005, 337, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Yutzy, W.H.; Fallon, R.; Inglis, S.; Lowy, D.R.; Schiller, J.T. Minor capsid protein of human genital papillomaviruses contains subdominant, cross-neutralizing epitopes. Virology 2000, 270, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Ishii, Y.; Ochi, H.; Matsumoto, T.; Yoshikawa, H.; Kanda, T. Neutralization of HPV16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the HPV16 minor capsid protein L2 surface region. Virology 2007, 358, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.T.; Schellenbacher, C.; Chackerian, B.; Roden, R.B. Progress and prospects for L2-based human papillomavirus vaccines. Expert Rev. Vaccines 2016, 15, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Lambert, P.F.; Ahlquist, P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 9311–9316. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004, 78, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, S.C.; Patterson, N.A.; Ozbun, M.A.; Lambert, P.F. The minor capsid protein L2 contributes to two steps in the human papillomavirus type 31 life cycle. J. Virol. 2005, 79, 3938–3948. [Google Scholar] [CrossRef] [PubMed]
- Volpers, C.; Schirmacher, P.; Streeck, R.E.; Sapp, M. Assembly of the major and the minor capsid protein of human papillomavirus type 33 into virus-like particles and tubular structures in insect cells. Virology 1994, 200, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; Meyers, C. Differential dependence on host cell glycosaminoglycans for infection of epithelial cells by high-risk HPV types. PLoS ONE 2013, 8, e68379. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Orth, G.; Breitburd, F.; Favre, M. Evidence for antigenic determinants shared by the structural polypeptides of (Shope) rabbit papillomavirus and human papillomavirus type 1. Virology 1978, 91, 243–255. [Google Scholar] [CrossRef]
- Rippe, R.A.; Meinke, W.J. Identification and characterization of the BPV-2 L2 protein. Virology 1989, 171, 298–301. [Google Scholar] [CrossRef]
- Favre, M. Structural polypeptides of rabbit, bovine, and human papillomaviruses. J. Virol. 1975, 15, 1239–12347. [Google Scholar] [PubMed]
- Hagensee, M.E.; Yaegashi, N.; Galloway, D.A. Self-assembly of human papillomavirus type 1 capsids by expression of the L1 protein alone or by coexpression of the L1 and L2 capsid proteins. J. Virol. 1993, 67, 315–322. [Google Scholar] [PubMed]
- Hagensee, M.E.; Olson, N.H.; Baker, T.S.; Galloway, D.A. Three-dimensional structure of vaccinia virus-produced human papillomavirus type 1 capsids. J. Virol. 1994, 68, 4503–4505. [Google Scholar] [PubMed]
- Belnap, D.M.; Olson, N.H.; Cladel, N.M.; Newcomb, W.W.; Brown, J.C.; Kreider, J.W.; Christensen, N.D.; Baker, T.S. Conserved features in papillomavirus and polyomavirus capsids. J. Mol. Biol. 1996, 259, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Trus, B.L.; Roden, R.B.; Greenstone, H.L.; Vrhel, M.; Schiller, J.T.; Booy, F.P. Novel structural features of bovine papillomavirus capsid revealed by a three-dimensional reconstruction to 9 A resolution. Nat. Struct. Biol. 1997, 4, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the papillomavirus capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Trus, B.L. The papillomavirus virion: A machine built to hide molecular Achilles’ heels. Adv. Exp. Med. Biol. 2012, 726, 403–422. [Google Scholar] [PubMed]
- Schellenbacher, C.; Roden, R.B.; Kirnbauer, R. Developments in L2-based human papillomavirus (HPV) vaccines. Virus Res. 2017, 231, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Rubio, I.; Seitz, H.; Canali, E.; Sehr, P.; Bolchi, A.; Tommasino, M.; Ottonello, S.; Muller, M. The N-terminal region of the human papillomavirus L2 protein contains overlapping binding sites for neutralizing, cross-neutralizing and non-neutralizing antibodies. Virology 2011, 409, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Jagu, S.; Karanam, B.; Wang, J.W.; Zayed, H.; Weghofer, M.; Brendle, S.A.; Balogh, K.K.; Tossi, K.P.; Roden, R.B.; Christensen, N.D. Durable immunity to oncogenic human papillomaviruses elicited by adjuvanted recombinant Adeno-associated virus-like particle immunogen displaying L2 17–36 epitopes. Vaccine 2015, 33, 5553–5563. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.D.; Dillner, J.; Eklund, C.; Carter, J.J.; Wipf, G.C.; Reed, C.A.; Cladel, N.M.; Galloway, D.A. Surface conformational and linear epitopes on HPV-16 and HPV-18 L1 virus-like particles as defined by monoclonal antibodies. Virology 1996, 223, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Spira, G.; Scharff, M.D. Identification of rare immunoglobulin switch variants using the ELISA spot assay. J. Immunol. Methods 1992, 148, 121–129. [Google Scholar] [CrossRef]
- Buck, C.B.; Thompson, C.D.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Maturation of papillomavirus capsids. J. Virol. 2005, 79, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Brendle, S.A.; Culp, T.D.; Broutian, T.R.; Christensen, N.D. Binding and neutralization characteristics of a panel of monoclonal antibodies to human papillomavirus 58. J. Gen. Virol. 2010, 91, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Mejia, A.F.; Culp, T.D.; Cladel, N.M.; Balogh, K.K.; Budgeon, L.R.; Buck, C.B.; Christensen, N.D. Preclinical model to test human papillomavirus virus (HPV) capsid vaccines in vivo using infectious HPV/cottontail rabbit papillomavirus chimeric papillomavirus particles. J. Virol. 2006, 80, 12393–12397. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Mayer, T.J.; Ozbun, M.A. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 1997, 71, 7381–7386. [Google Scholar] [PubMed]
- Conway, M.J.; Alam, S.; Christensen, N.D.; Meyers, C. Overlapping and independent structural roles for human papillomavirus type 16 L2 conserved cysteines. Virology 2009, 393, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Alam, S.; Ryndock, E.J.; Cruz, L.; Christensen, N.D.; Roden, R.B.; Meyers, C. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J. Virol. 2009, 83, 10515–10526. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Cruz, L.; Alam, S.; Christensen, N.D.; Meyers, C. Differentiation-dependent interpentameric disulfide bond stabilizes native human papillomavirus type 16. PLoS ONE 2011, 6, e22427. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Bywaters, S.M.; Brendle, S.A.; Lee, H.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Christensen, N.D.; Hafenstein, S. Structural comparison of four different antibodies interacting with human papillomavirus 16 and mechanisms of neutralization. Virology 2015, 483, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Bywaters, S.M.; Brendle, S.A.; Lee, H.; Ashley, R.E.; Christensen, N.D.; Hafenstein, S. The U4 Antibody Epitope on Human Papillomavirus 16 Identified by Cryo-electron Microscopy. J. Virol. 2015, 89, 12108–12117. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Brendle, S.A.; Bywaters, S.M.; Guan, J.; Ashley, R.E.; Yoder, J.D.; Makhov, A.M.; Conway, J.F.; Christensen, N.D.; Hafenstein, S. A cryo-electron microscopy study identifies the complete H16.V5 epitope and reveals global conformational changes initiated by binding of the neutralizing antibody fragment. J. Virol. 2015, 89, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Raifu, M.; Howard, M.; Smith, L.; Hansen, D.; Goldsby, R.; Ratner, D. Universal PCR amplification of mouse immunoglobulin gene variable regions: The design of degenerate primers and an assessment of the effect of DNA polymerase 3′ to 5′ exonuclease activity. J. Immunol. Methods 2000, 233, 167–177. [Google Scholar] [CrossRef]
- Hu, J.; Peng, X.; Schell, T.D.; Budgeon, L.R.; Cladel, N.M.; Christensen, N.D. An HLA-A2.1-transgenic rabbit model to study immunity to papillomavirus infection. J. Immunol. 2006, 177, 8037–8045. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Christensen, N.D. Quantitative RT-PCR assay for HPV infection in cultured cells. J. Virol. Methods 2003, 111, 135–144. [Google Scholar] [CrossRef]
- Hu, J.; Budgeon, L.R.; Cladel, N.M.; Culp, T.D.; Balogh, K.K.; Christensen, N.D. Detection of L1, infectious virions and anti-L1 antibody in domestic rabbits infected with cottontail rabbit papillomavirus. J. Gen. Virol. 2007, 88, 3286–3293. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Tang, F.; Yang, C.; Zhang, W.; Berguist, J.; Wang, B.; Mi, J.; Zhang, J. Quantitative dot blot analysis (QDB), a versatile high throughput immunoblot method. Oncotarget 2017, 8, 58553–58562. [Google Scholar] [CrossRef] [PubMed]
- Brochet, X.; Lefranc, M.P.; Giudicelli, V. IMGT/V-QUEST: The highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008, 36, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Giudicelli, V.; Brochet, X.; Lefranc, M.P. IMGT/V-QUEST: IMGT standardized analysis of the im.unoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc. 2011, 2011, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Selinka, H.C.; Giroglou, T.; Nowak, T.; Christensen, N.D.; Sapp, M. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 2003, 77, 12961–12967. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Gambhira, R.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. Mechanisms of human papillomavirus type 16 neutralization by l2 cross-neutralizing and l1 type-specific antibodies. J. Virol. 2008, 82, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Schiller, J.T. The role of furin in papillomavirus infection. Future Microbiol. 2009, 4, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.D.; Cladel, N.M.; Reed, C.A.; Budgeon, L.R.; Embers, M.E.; Skulsky, D.M.; McClements, W.L.; Ludmerer, S.W.; Jansen, K.U. Hybrid papillomavirus L1 molecules assemble into virus-like particles that reconstitute conformational epitopes and induce neutralizing antibodies to distinct HPV types. Virology 2001, 291, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Garcea, R.L.; Grigorieff, N.; Harrison, S.C. Subunit interactions in bovine papillomavirus. Proc. Natl. Acad. Sci. USA 2010, 107, 6298–6303. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.L.; Dore, I.; Porta, C.; van Regenmortel, M.H. Conformational specificity of monoclonal antibodies used in the diagnosis of tomato mosaic virus. Arch. Virol. 1987, 94, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Mierendorf, R.C., Jr.; Dimond, R.L. Functional heterogeneity of monoclonal antibodies obtained using different screening assays. Anal. Biochem. 1983, 135, 221–229. [Google Scholar] [CrossRef]
- McCullough, K.C.; Crowther, J.R.; Butcher, R.N. Alteration in antibody reactivity with foot-and-mouth disease virus (FMDV) 146S antigen before and after binding to a solid phase or complexing with specific antibody. J. Immunol. Methods 1985, 82, 91–100. [Google Scholar] [CrossRef]
- Bruck, C.; Portetelle, D.; Burny, A.; Zavada, J. Topographical analysis by monoclonal antibodies of BLV-gp51 epitopes involved in viral functions. Virology 1982, 122, 353–362. [Google Scholar] [CrossRef]
- Al Moudallal, Z.; Briand, J.P.; Van Regenmortel, M.H. Monoclonal antibodies as probes of the antigenic structure of tobacco mosaic virus. EMBO J. 1982, 1, 1005–1010. [Google Scholar] [PubMed]
- Al Moudallal, Z.; Altschuh, D.; Briand, J.P.; Van Regenmortel, M.H. Comparative sensitivity of different ELISA methods for detecting monoclonal antibodies to viruses. Dev. Biol. Stand. 1984, 57, 35–40. [Google Scholar] [PubMed]
- Dekker, E.L.; Porta, C.; Van Regenmortel, M.H.V. Limitations of different ELISA procedures for localizing epitopes in viral coat protein subunits. Arch. Virol. 1989, 105, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Meloen, R.H.; Briaire, J. A study of the cross-reacting antigens on the intact foot-and-mouth disease virus and its 12S Subunits with antisera against the structural proteins. J. Gen. Virol. 1980, 51, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Meloen, R.H.; Rowlands, D.J.; Brown, F. Comparison of the antibodies elicited by the individual structural polypeptides of foot-and mouth disease and polio viruses. J. Gen. Virol. 1979, 45, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Wilson, J.E. A modified ELISA that selectively detects monoclonal antibodies recognizing native antigen. J. Immunol. Methods 1986, 94, 31–35. [Google Scholar] [CrossRef]
- Vaidya, H.C.; Dietzler, D.N.; Ladenson, J.H. Inadequacy of traditional ELISA for screening hybridoma supernatants for murine monoclonal antibodies. Hybrid 1985, 4, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Djavadi-Ohaniance, L.; Friguet, B.; Goldberg, M.E. Structural and functional influence of enzyme-antibody interactions: Effects of eight different monoclonal antibodies on the enzymatic activity of Escherichia coli tryptophan synthase. Biochemistry 1984, 23, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Buck, C.B.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J. Virol. 2007, 81, 8784–8792. [Google Scholar] [CrossRef] [PubMed]
- White, W.I.; Wilson, S.D.; Palmer-Hill, F.J.; Woods, R.M.; Ghim, S.J.; Hewitt, L.A.; Goldman, D.M.; Burke, S.J.; Jenson, A.B.; Koenig, S.; et al. Characterization of a major neutralizing epitope on human papillomavirus type 16 L1. J. Virol. 1999, 73, 4882–4889. [Google Scholar] [PubMed]
- Conway, M.J.; Cruz, L.; Alam, S.; Christensen, N.D.; Meyers, C. Cross-neutralization potential of native human papillomavirus N-terminal L2 epitopes. PLoS ONE 2011, 6, e16405. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Spatz, C.M.; Reed, C.A.; Christensen, N.D. Binding and neutralization efficiencies of monoclonal antibodies, Fab fragments, and scFv specific for L1 epitopes on the capsid of infectious HPV particles. Virology 2007, 361, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Bywaters, S.M.; Brendle, S.A.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Christensen, N.D.; Hafenstein, S. Cryoelectron Microscopy Maps of Human Papillomavirus 16 Reveal L2 Densities and Heparin Binding Site. Structure 2017, 25, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Bronnimann, M.P.; Calton, C.M.; Chiquette, S.F.; Li, S.; Lu, M.; Chapman, J.A.; Bratton, K.N.; Schlegel, A.M.; Campos, S.K. Furin Cleavage of L2 during Papillomavirus Infection: Minimal Dependence on Cyclophilins. J. Virol. 2016, 90, 6224–6234. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D. Production of papillomavirus-based gene transfer vectors. Curr. Protoc. Cell Biol. 2007. [Google Scholar] [CrossRef]
- Gambhira, R.; Jagu, S.; Karanam, B.; Gravitt, P.E.; Culp, T.D.; Christensen, N.D.; Roden, R.B. Protection of rabbits against challenge with rabbit papillomaviruses by immunization with the N terminus of human papillomavirus type 16 minor capsid antigen L2. J. Virol. 2007, 81, 11585–11592. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
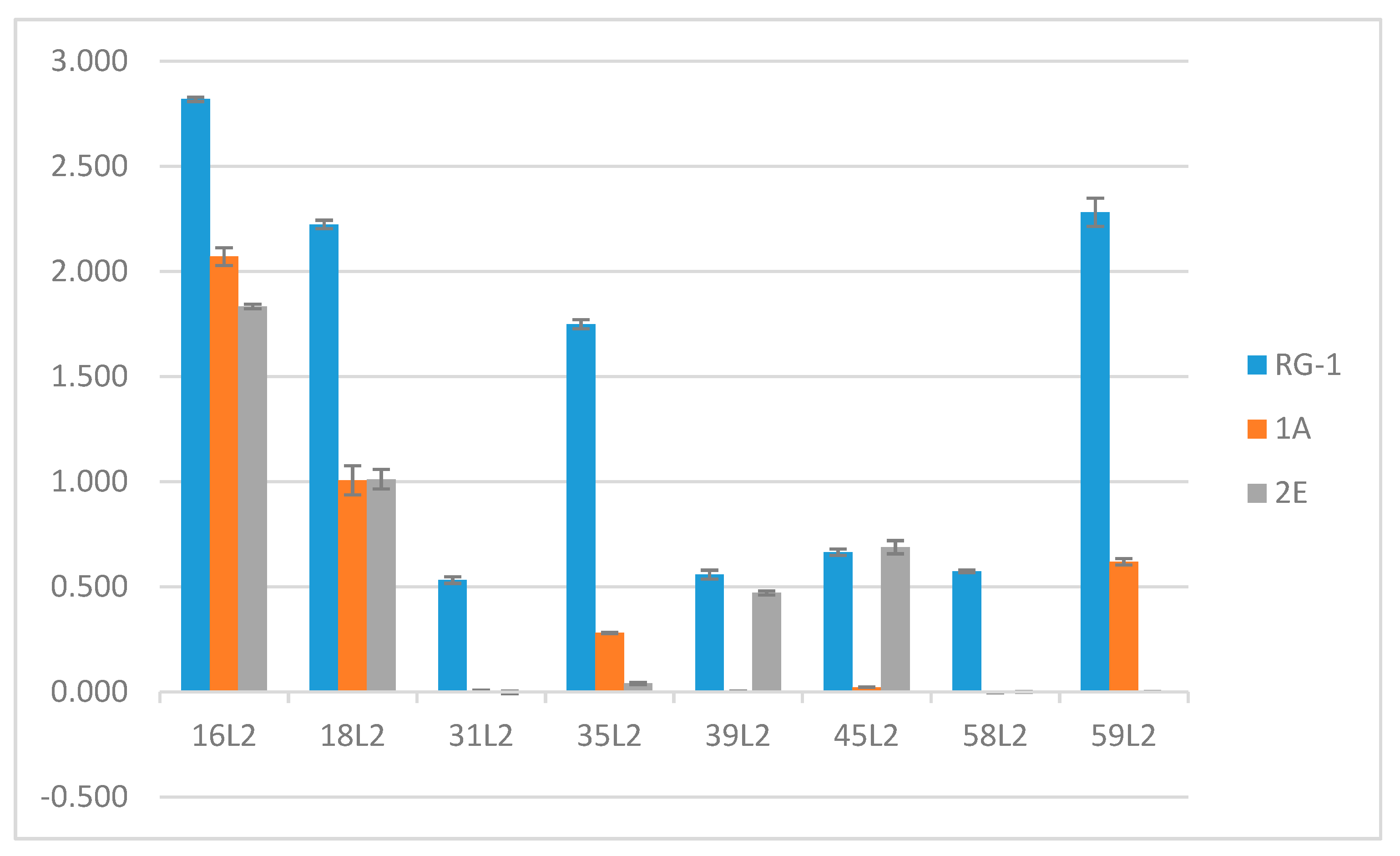{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide ELISA | IF | |||||
|---|---|---|---|---|---|---|
| Peptide Sequence aa17—36 | RG-1 | H16.L2.1A | H16.L2.2E | H16.L2.1A | H16.L2.2E | |
| HPV16 L2 | QLYKTCKQAGTCPPDIIPKV | ++++ | ++++ | ++++ | + | + |
| HPV18 L2 | DLYKTCKQSGTCPPDVVPKV | ++++ | ++ | ++ | + | + |
| HPV31 L2 | QLYQTCKAAGTCPSDVIPKI | + | − | − | NT | NT |
| HPV35 L2 | QLYRTCKAAGTCPPDVIPKV | +++ | + | − | ± | − |
| HPV39 L2 | DLYRTCKQSGTCPPDVVDKV | + | − | + | − | + |
| HPV45 L2 | DLYRTCKQSGTCPPDVINKV | + | − | + | − | + |
| HPV58 L2 | QLYQTCKASGTCPPDVIPKV | + | − | − | NT | NT |
| HPV59 L2 | DLYKTCKQAGTCPSDVINKV | ++++ | + | − | + | − |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bywaters, S.M.; Brendle, S.A.; Tossi, K.P.; Biryukov, J.; Meyers, C.; Christensen, N.D. Antibody Competition Reveals Surface Location of HPV L2 Minor Capsid Protein Residues 17–36. Viruses 2017, 9, 336. https://doi.org/10.3390/v9110336
Bywaters SM, Brendle SA, Tossi KP, Biryukov J, Meyers C, Christensen ND. Antibody Competition Reveals Surface Location of HPV L2 Minor Capsid Protein Residues 17–36. Viruses. 2017; 9(11):336. https://doi.org/10.3390/v9110336
Chicago/Turabian StyleBywaters, Stephanie M., Sarah A. Brendle, Kerstin P. Tossi, Jennifer Biryukov, Craig Meyers, and Neil D. Christensen. 2017. "Antibody Competition Reveals Surface Location of HPV L2 Minor Capsid Protein Residues 17–36" Viruses 9, no. 11: 336. https://doi.org/10.3390/v9110336
APA StyleBywaters, S. M., Brendle, S. A., Tossi, K. P., Biryukov, J., Meyers, C., & Christensen, N. D. (2017). Antibody Competition Reveals Surface Location of HPV L2 Minor Capsid Protein Residues 17–36. Viruses, 9(11), 336. https://doi.org/10.3390/v9110336