Characterization of HIV-1 Near Full-Length Proviral Genome Quasispecies from Patients with Undetectable Viral Load Undergoing First-Line HAART Therapy

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Sample Collection
2.2. DNA Extraction and PCR of Proviral DNA
2.3. Library Construction and NGS
2.4. Data Analysis
2.4.1. Analysis of Resistance Mutations
2.4.2. Phylogenetic Analysis
2.4.3. Analysis of Viral Tropism
3. Results
4. Discussion
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). UNAIDS Data 2017; UNAIDS: Geneva, Switzerland, 2017. [Google Scholar]
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). Ending AIDS: Progress Towards the 90-90-90 Targets; UNAIDS: Geneva, Switzerland, 2017. [Google Scholar]
- Brazilian Ministry of Health. Clinical Protocol and Therapeutic Guidelines for the Management of HIV Infection in Adults; Department of Surveillance, Prevention and Control of Infections Sexually Tramsitted, HIV/AIDS and Viral Hepatites: Brasilia, Brazil, 2017.
- Brazilian Ministry of Health. Clinical Protocol and Therapeutic Guidelines for Management of HIV Infection in Adults; Department of Surveillance, Prevention and Control of Infections Sexually Tramsitted, HIV/AIDS and Viral Hepatites: Brasilia, Brazil, 2013.
- Roberts, J.D.; Bebenek, K.; Kunkel, T.A. The accuracy of reverse transcriptase from HIV-1. Science 1988, 242, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Xu, H.; Wainberg, M.A. Defective HIV-1 quasispecies in the form of multiply drug-resistant proviral DNA within cells can be rescued by superinfection with different subtype variants of HIV-1 and by HIV-2 and SIV. J. Antimicrob. Chemother. 2014, 69, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Wain-Hobson, S. The fastest genome evolution ever described: HIV variation in situ. Curr. Opin. Genet. Dev. 1993, 3, 878–883. [Google Scholar] [CrossRef]
- Overbaugh, J.; Bangham, C.R. Selection forces and constraints on retroviral sequence variation. Science 2001, 292, 1106–1109. [Google Scholar] [CrossRef] [PubMed]
- Atlas, A.; Granath, F.; Lindstrom, A.; Lidman, K.; Lindback, S.; Alaeus, A. Impact of HIV type 1 genetic subtype on the outcome of antiretroviral therapy. AIDS Res. Hum. Retrovir. 2005, 21, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.W.; Shafer, R.W. HIV-1 antiretroviral resistance: Scientific principles and clinical applications. Drugs 2012, 72, e1–e25. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.E.; Camacho, R.J.; Otelea, D.; Kuritzkes, D.R.; Fleury, H.; Kiuchi, M.; Heneine, W.; Kantor, R.; Jordan, M.R.; Schapiro, J.M.; et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS ONE 2009, 4, e4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafer, R.W.; Rhee, S.Y.; Pillay, D.; Miller, V.; Sandstrom, P.; Schapiro, J.M.; Kuritzkes, D.R.; Bennett, D. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS 2007, 21, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Castor, D.; Low, A.; Evering, T.; Karmon, S.; Davis, B.; Figueroa, A.; LaMar, M.; Garmon, D.; Mehandru, S.; Markowitz, M. Transmitted drug resistance and phylogenetic relationships among acute and early HIV-1-infected individuals in New York City. J. Acquir. Immune Defic. Syndr. 2012, 61, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.; Bannert, N.; Sonnerborg, A.; de Mendoza, C.; Price, M.; Zangerle, R.; Chaix, M.L.; Prins, M.; Kran, A.B.; Gill, J.; et al. Temporal trends of transmitted HIV drug resistance in a multinational seroconversion cohort. AIDS 2017. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, S.J.; Ssempijja, V.; Galiwango, R.; Ndyanabo, A.; Nakigozi, G.; Lyagoba, F.; Nazziwa, J.; Redd, A.; Lamers, S.L.; Gray, R.; et al. Low rates of transmitted drug resistance among newly identified HIV-1 seroconverters in Rural Rakai, Uganda. AIDS Res. Hum. Retrovir. 2017, 33, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, L.; Sun, M.; Sun, J.; Lu, H. An analysis of drug resistance among people living with HIV/AIDS in Shanghai, China. PLoS ONE 2017, 12, e0165110. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, B.; Trovao, N.S.; Baele, G.; van Wijngaerden, E.; Vandamme, A.M.; van Laethem, K.; Lemey, P. Quantifying next generation sequencing sample pre-processing bias in HIV-1 complete genome sequencing. Viruses 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, R.M.; Kuritzkes, D.R.; Johnson, V.A.; Mellors, J.W.; Sullivan, J.L.; Swanstrom, R.; D’Aquila, R.T.; van Gorder, M.; Holodniy, M.; Lloyd, R.M., Jr.; et al. Accuracy of the TRUGENE HIV-1 genotyping kit. J. Clin. Microbiol. 2003, 41, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Halvas, E.K.; Aldrovandi, G.M.; Balfe, P.; Beck, I.A.; Boltz, V.F.; Coffin, J.M.; Frenkel, L.M.; Hazelwood, J.D.; Johnson, V.A.; Kearney, M.; et al. Blinded, multicenter comparison of methods to detect a drug-resistant mutant of human immunodeficiency virus type 1 at low frequency. J. Clin. Microbiol. 2006, 44, 2612–2614. [Google Scholar] [CrossRef] [PubMed]
- Larder, B.A.; Kohli, A.; Kellam, P.; Kemp, S.D.; Kronick, M.; Henfrey, R.D. Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing. Nature 1993, 365, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Bellecave, P.; Recordon-Pinson, P.; Papuchon, J.; Vandenhende, M.A.; Reigadas, S.; Tauzin, B.; Fleury, H. Detection of low-frequency HIV type 1 reverse transcriptase drug resistance mutations by ultradeep sequencing in naive HIV type 1-infected individuals. AIDS Res. Hum. Retrovir. 2014, 30, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.M.; Chin, E.N.; Bimber, B.N.; Sanabani, S.S.; Tarosso, L.F.; Costa, P.R.; Sauer, M.M.; Kallas, E.G.; O’Connor, D.H. Low-cost ultra-wide genotyping using Roche/454 pyrosequencing for surveillance of HIV drug resistance. PLoS ONE 2012, 7, e36494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, S.; Tully, D.C.; Gondwe, C.; Wood, C. Low-abundance resistant mutations in HIV-1 subtype C antiretroviral therapy-naive individuals as revealed by pyrosequencing. Curr. HIV Res. 2013, 11, 43–49. [Google Scholar] [PubMed]
- Ji, H.; Li, Y.; Graham, M.; Liang, B.B.; Pilon, R.; Tyson, S.; Peters, G.; Tyler, S.; Merks, H.; Bertagnolio, S.; et al. Next-generation sequencing of dried blood spot specimens: A novel approach to HIV drug-resistance surveillance. Antivir. Ther. 2011, 16, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Simen, B.B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; Macarthur, R.D.; Baxter, J.D.; Huang, C.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Terry Beirn Community Programs for Clinical Research on, A. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 2009, 199, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Cane, P.A. New developments in HIV drug resistance. J. Antimicrob. Chemother. 2009, 64 (Suppl. 1), i37–i40. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Metzner, K.J.; Rauch, P.; Walter, H.; Boesecke, C.; Zollner, B.; Jessen, H.; Schewe, K.; Fenske, S.; Gellermann, H.; Stellbrink, H.J. Detection of minor populations of drug-resistant HIV-1 in acute seroconverters. AIDS 2005, 19, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Ghosn, J.; Pellegrin, I.; Goujard, C.; Deveau, C.; Viard, J.P.; Galimand, J.; Harzic, M.; Tamalet, C.; Meyer, L.; Rouzioux, C.; et al. HIV-1 resistant strains acquired at the time of primary infection massively fuel the cellular reservoir and persist for lengthy periods of time. AIDS 2006, 20, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Booth, C.L.; Geretti, A.M. Prevalence and determinants of transmitted antiretroviral drug resistance in HIV-1 infection. J. Antimicrob. Chemother. 2007, 59, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Pingen, M.; Nijhuis, M.; de Bruijn, J.A.; Boucher, C.A.; Wensing, A.M. Evolutionary pathways of transmitted drug-resistant HIV-1. J. Antimicrob. Chemother. 2011, 66, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Kyeyune, F.; Gibson, R.M.; Nankya, I.; Venner, C.; Metha, S.; Akao, J.; Ndashimye, E.; Kityo, C.M.; Salata, R.A.; Mugyenyi, P.; et al. Low-Frequency drug resistance in HIV-infected ugandans on antiretroviral treatment is associated with regimen failure. Antimicrob. Agents Chemother. 2016, 60, 3380–3397. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, M.; Chiarella, J.; Yang, R.; Schnittman, S.; Wirtz, V.; Uy, J.; Seekins, D.; Krystal, M.; Mancini, M.; McGrath, D.; et al. Prevalence and clinical significance of HIV drug resistance mutations by ultra-deep sequencing in antiretroviral-naive subjects in the CASTLE study. PLoS ONE 2010, 5, e10952. [Google Scholar] [CrossRef] [PubMed]
- Vandenhende, M.A.; Bellecave, P.; Recordon-Pinson, P.; Reigadas, S.; Bidet, Y.; Bruyand, M.; Bonnet, F.; Lazaro, E.; Neau, D.; Fleury, H.; et al. Prevalence and evolution of low frequency HIV drug resistance mutations detected by ultra deep sequencing in patients experiencing first line antiretroviral therapy failure. PLoS ONE 2014, 9, e86771. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Matsuda, M.; Hattori, J.; Shiino, T.; Matano, T.; Heneine, W.; Johnson, J.A.; Sugiura, W. Longitudinal detection and persistence of minority drug-resistant populations and their effect on salvage therapy. PLoS ONE 2015, 10, e0135941. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Li, J.F.; Wei, X.; Lipscomb, J.; Irlbeck, D.; Craig, C.; Smith, A.; Bennett, D.E.; Monsour, M.; Sandstrom, P.; et al. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naive populations and associate with reduced treatment efficacy. PLoS Med. 2008, 5, e158. [Google Scholar] [CrossRef] [PubMed]
- Pingen, M.; van der Ende, M.E.; Wensing, A.M.; El Barzouhi, A.; Simen, B.B.; Schutten, M.; Boucher, C.A. Deep sequencing does not reveal additional transmitted mutations in patients diagnosed with HIV-1 variants with single nucleoside reverse transcriptase inhibitor resistance mutations. HIV Med. 2013, 14, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Peuchant, O.; Thiebaut, R.; Capdepont, S.; Lavignolle-Aurillac, V.; Neau, D.; Morlat, P.; Dabis, F.; Fleury, H.; Masquelier, B.; Cohort, A.C.A. Transmission of HIV-1 minority-resistant variants and response to first-line antiretroviral therapy. AIDS 2008, 22, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Boltz, V.F.; Ambrose, Z.; Kearney, M.F.; Shao, W.; Kewalramani, V.N.; Maldarelli, F.; Mellors, J.W.; Coffin, J.M. Ultrasensitive allele-specific PCR reveals rare preexisting drug-resistant variants and a large replicating virus population in macaques infected with a simian immunodeficiency virus containing human immunodeficiency virus reverse transcriptase. J. Virol. 2012, 86, 12525–12530. [Google Scholar] [CrossRef] [PubMed]
- Metzner, K.J.; Rauch, P.; von Wyl, V.; Leemann, C.; Grube, C.; Kuster, H.; Boni, J.; Weber, R.; Gunthard, H.F. Efficient suppression of minority drug-resistant HIV type 1 (HIV-1) variants present at primary HIV-1 infection by ritonavir-boosted protease inhibitor-containing antiretroviral therapy. J. Infect. Dis. 2010, 201, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianella, S.; Delport, W.; Pacold, M.E.; Young, J.A.; Choi, J.Y.; Little, S.J.; Richman, D.D.; Kosakovsky Pond, S.L.; Smith, D.M. Detection of minority resistance during early HIV-1 infection: Natural variation and spurious detection rather than transmission and evolution of multiple viral variants. J. Virol. 2011, 85, 8359–8367. [Google Scholar] [CrossRef] [PubMed]
- Stekler, J.D.; Ellis, G.M.; Carlsson, J.; Eilers, B.; Holte, S.; Maenza, J.; Stevens, C.E.; Collier, A.C.; Frenkel, L.M. Prevalence and impact of minority variant drug resistance mutations in primary HIV-1 infection. PLoS ONE 2011, 6, e28952. [Google Scholar] [CrossRef] [PubMed]
- Lataillade, M.; Chiarella, J.; Yang, R.; DeGrosky, M.; Uy, J.; Seekins, D.; Simen, B.; St John, E.; Moreno, E.; Kozal, M. Virologic failures on initial boosted-PI regimen infrequently possess low-level variants with major PI resistance mutations by ultra-deep sequencing. PLoS ONE 2012, 7, e30118. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, C.; Lee, G.Q.; Rodriguez, C.; Visseaux, B.; Storto, A.; Fagard, C.; Molina, J.M.; Katlama, C.; Yazdanpanah, Y.; Harrigan, P.R.; et al. Highly frequent HIV-1 minority resistant variants at baseline of the ANRS 139 TRIO trial had a limited impact on virological response. J. Antimicrob. Chemother. 2015, 70, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Hare, C.B.; Mellors, J.; Krambrink, A.; Su, Z.; Skiest, D.; Margolis, D.M.; Patel, S.S.; Barnas, D.; Frenkel, L.; Coombs, R.W.; et al. Detection of nonnucleoside reverse-transcriptase inhibitor-resistant HIV-1 after discontinuation of virologically suppressive antiretroviral therapy. Clin. Infect. Dis. 2008, 47, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Chiarella, J.; Simen, B.B.; Hanczaruk, B.; Egholm, M.; Landry, M.L.; Dieckhaus, K.; Rosen, M.I.; Kozal, M.J. Low-abundance HIV drug-resistant viral variants in treatment-experienced persons correlate with historical antiretroviral use. PLoS ONE 2009, 4, e6079. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Paredes, R.; Ribaudo, H.J.; Svarovskaia, E.S.; Metzner, K.J.; Kozal, M.J.; Hullsiek, K.H.; Balduin, M.; Jakobsen, M.R.; Geretti, A.M.; et al. Low-frequency HIV-1 drug resistance mutations and risk of NNRTI-based antiretroviral treatment failure: A systematic review and pooled analysis. JAMA 2011, 305, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Cozzi-Lepri, A.; Noguera-Julian, M.; di Giallonardo, F.; Schuurman, R.; Daumer, M.; Aitken, S.; Ceccherini-Silberstein, F.; D’Arminio Monforte, A.; Geretti, A.M.; Booth, C.L.; et al. Low-frequency drug-resistant HIV-1 and risk of virological failure to first-line NNRTI-based ART: A multicohort European case-control study using centralized ultrasensitive 454 pyrosequencing. J. Antimicrob. Chemother. 2015, 70, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Paredes, R.; Lalama, C.M.; Ribaudo, H.J.; Schackman, B.R.; Shikuma, C.; Giguel, F.; Meyer, W.A., 3rd; Johnson, V.A.; Fiscus, S.A.; D’Aquila, R.T.; et al. Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. J. Infect. Dis. 2010, 201, 662–671. [Google Scholar] [PubMed]
- Papuchon, J.; Pinson, P.; Lazaro, E.; Reigadas, S.; Guidicelli, G.; Taupin, J.L.; Neau, D.; Fleury, H.; The Provir/Latitude 45 Project. Resistance mutations and CTL epitopes in archived HIV-1 DNA of patients on antiviral treatment: Toward a new concept of vaccine. PLoS ONE 2013, 8, e69029. [Google Scholar] [CrossRef] [PubMed]
- CDC. Revised surveillance case definition for HIV infection—United States, 2014. Morb. Mortal. Wkly. Rep. 2014, 63, 1–10. [Google Scholar]
- Sanabani, S.; Neto, W.K.; de Sa Filho, D.J.; Diaz, R.S.; Munerato, P.; Janini, L.M.; Sabino, E.C. Full-length genome analysis of human immunodeficiency virus type 1 subtype C in Brazil. AIDS Res. Hum. Retrovir. 2006, 22, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Ode, H.; Matsuda, M.; Matsuoka, K.; Hachiya, A.; Hattori, J.; Kito, Y.; Yokomaku, Y.; Iwatani, Y.; Sugiura, W. Quasispecies Analyses of the HIV-1 Near-full-length Genome With Illumina MiSeq. Front. Microbiol. 2015, 6, 1258. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.M.; Bailey, A.L.; Mehta, S.H.; Hughes, A.L.; Kirk, G.D.; Westergaard, R.P.; O’Connor, D.H. Cross-clade simultaneous HIV drug resistance genotyping for reverse transcriptase, protease, and integrase inhibitor mutations by Illumina MiSeq. Retrovirology 2014, 11, 122. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; Calvez, V.; Gunthard, H.F.; Johnson, V.A.; Paredes, R.; Pillay, D.; Shafer, R.W.; Richman, D.D. 2015 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2015, 23, 132–141. [Google Scholar] [PubMed]
- Brehm, J.H.; Koontz, D.; Meteer, J.D.; Pathak, V.; Sluis-Cremer, N.; Mellors, J.W. Selection of mutations in the connection and RNase H domains of human immunodeficiency virus type 1 reverse transcriptase that increase resistance to 3′-azido-3′-dideoxythymidine. J. Virol. 2007, 81, 7852–7859. [Google Scholar] [CrossRef] [PubMed]
- Dau, B.; Ayers, D.; Singer, J.; Harrigan, P.R.; Brown, S.; Kyriakides, T.; Cameron, D.W.; Angus, B.; Holodniy, M. Connection domain mutations in treatment-experienced patients in the OPTIMA trial. J. Acquir. Immune Defic. Syndr. 2010, 54, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Maldarelli, F.; Hase, S.; Takebe, Y.; Pathak, V.K. Subtype-specific differences in the human immunodeficiency virus type 1 reverse transcriptase connection subdomain of CRF01_AE are associated with higher levels of resistance to 3′-azido-3′-deoxythymidine. J. Virol. 2009, 83, 8502–8513. [Google Scholar] [CrossRef] [PubMed]
- Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Pathak, V.K. The “Connection” Between HIV Drug Resistance and RNase H. Viruses 2010, 2, 1476–1503. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Vingerhoets, J.; Fransen, S.; Tambuyzer, L.; Azijn, H.; Frantzell, A.; Paredes, R.; Coakley, E.; Nijs, S.; Clotet, B.; et al. Connection domain mutations in HIV-1 reverse transcriptase do not impact etravirine susceptibility and virologic responses to etravirine-containing regimens. Antimicrob. Agents Chemother. 2011, 55, 2872–2879. [Google Scholar] [CrossRef] [PubMed]
- Lengruber, R.B.; Delviks-Frankenberry, K.A.; Nikolenko, G.N.; Baumann, J.; Santos, A.F.; Pathak, V.K.; Soares, M.A. Phenotypic characterization of drug resistance-associated mutations in HIV-1 RT connection and RNase H domains and their correlation with thymidine analogue mutations. J. Antimicrob. Chemother. 2011, 66, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Paredes, R.; Puertas, M.C.; Bannister, W.; Kisic, M.; Cozzi-Lepri, A.; Pou, C.; Bellido, R.; Betancor, G.; Bogner, J.; Gargalianos, P.; et al. A376S in the connection subdomain of HIV-1 reverse transcriptase confers increased risk of virological failure to nevirapine therapy. J. Infect. Dis. 2011, 204, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Lengruber, R.B.; Soares, E.A.; Jere, A.; Sprinz, E.; Martinez, A.M.; Silveira, J.; Sion, F.S.; Pathak, V.K.; Soares, M.A. Conservation patterns of HIV-1 RT connection and RNase H domains: Identification of new mutations in NRTI-treated patients. PLoS ONE 2008, 3, e1781. [Google Scholar] [CrossRef] [PubMed]
- Eshleman, S.H.; Hackett, J., Jr.; Swanson, P.; Cunningham, S.P.; Drews, B.; Brennan, C.; Devare, S.G.; Zekeng, L.; Kaptue, L.; Marlowe, N. Performance of the Celera Diagnostics ViroSeq HIV-1 Genotyping System for sequence-based analysis of diverse human immunodeficiency virus type 1 strains. J. Clin. Microbiol. 2004, 42, 2711–2717. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; McLnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [PubMed]
- Jeanne, N.; Saliou, A.; Carcenac, R.; Lefebvre, C.; Dubois, M.; Cazabat, M.; Nicot, F.; Loiseau, C.; Raymond, S.; Izopet, J.; et al. Position-specific automated processing of V3 env ultra-deep pyrosequencing data for predicting HIV-1 tropism. Sci. Rep. 2015, 5, 16944. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, T.; Sander, O.; Sierra, S.; Thielen, A.; Kaiser, R. Bioinformatics prediction of HIV coreceptor usage. Nat. Biotechnol. 2007, 25, 1407–1410. [Google Scholar] [CrossRef] [PubMed]
- Delgado, E.; Fernandez-Garcia, A.; Vega, Y.; Cuevas, T.; Pinilla, M.; Garcia, V.; Sanchez, M.; Gonzalez, M.; Sanchez, A.M.; Thomson, M.M.; et al. Evaluation of genotypic tropism prediction tests compared with in vitro co-receptor usage in HIV-1 primary isolates of diverse subtypes. J. Antimicrob. Chemother. 2012, 67, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.M.; Meyer, A.M.; Winner, D.; Archer, J.; Feyertag, F.; Ruiz-Mateos, E.; Leal, M.; Robertson, D.L.; Schmotzer, C.L.; Quinones-Mateu, M.E. Sensitive deep-sequencing-based HIV-1 genotyping assay to simultaneously determine susceptibility to protease, reverse transcriptase, integrase, and maturation inhibitors, as well as HIV-1 coreceptor tropism. Antimicrob. Agents Chemother. 2014, 58, 2167–2185. [Google Scholar] [CrossRef] [PubMed]
- Raymond, S.; Delobel, P.; Mavigner, M.; Ferradini, L.; Cazabat, M.; Souyris, C.; Sandres-Saune, K.; Pasquier, C.; Marchou, B.; Massip, P.; et al. Prediction of HIV type 1 subtype C tropism by genotypic algorithms built from subtype B viruses. J. Acquir. Immune Defic. Syndr. 2010, 53, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, L.P.; Wensing, A.M.; Kaiser, R.; Brun-Vezinet, F.; Clotet, B.; de Luca, A.; Dressler, S.; Garcia, F.; Geretti, A.M.; Klimkait, T.; et al. European Consensus Group on clinical management of tropism, t., European guidelines on the clinical management of HIV-1 tropism testing. Lancet Infect. Dis. 2011, 11, 394–407. [Google Scholar] [CrossRef]
- Kaleebu, P.; French, N.; Mahe, C.; Yirrell, D.; Watera, C.; Lyagoba, F.; Nakiyingi, J.; Rutebemberwa, A.; Morgan, D.; Weber, J.; et al. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J. Infect. Dis. 2002, 185, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Archary, D.; Gordon, M.L.; Green, T.N.; Coovadia, H.M.; Goulder, P.J.; Ndung’u, T. HIV-1 subtype C envelope characteristics associated with divergent rates of chronic disease progression. Retrovirology 2010, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Casado, C.; Colombo, S.; Rauch, A.; Martinez, R.; Gunthard, H.F.; Garcia, S.; Rodriguez, C.; del Romero, J.; Telenti, A.; Lopez-Galindez, C. Host and viral genetic correlates of clinical definitions of HIV-1 disease progression. PLoS ONE 2010, 5, e11079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, P.J.; Carrington, M. The impact of host genetic variation on infection with HIV-1. Nat. Immunol. 2015, 16, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Leite, T.C.; Campos, D.P.; Coelho, A.B.; Teixeira, S.L.; Veloso, V.; Morgado, M.G.; Guimaraes, M.L. Impact of HIV-1 subtypes on AIDS progression in a Brazilian cohort. AIDS Res. Hum. Retrovir. 2017, 33, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Osmanov, S.; Pattou, C.; Walker, N.; Schwardlander, B.; Esparza, J.; WHO-UNAIDS Network for HIV Isolation and Characterization. Estimated global distribution and regional spread of HIV-1 genetic subtypes in the year 2000. J. Acquir. Immune Defic. Syndr. 2002, 29, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S.; WHO-UNAIDS Network for HIV Isolation and Characterisation. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 2011, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, D.M.; Almeida, S.E. HIV-1 subtype B: Traces of a pandemic. Virology 2016, 495, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Schrago, C.G.; Martinez, A.M.; Mendoza-Sassi, R.; Silveira, J.; Sousa, T.M.; Lengruber, R.B.; Soares, E.A.; Sprinz, E.; Soares, M.A. Epidemiologic and evolutionary trends of HIV-1 CRF31_BC-related strains in southern Brazil. J. Acquir. Immune Defic. Syndr. 2007, 45, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Passaes, C.P.; Guimaraes, M.L.; Bello, G.; Morgado, M.G. Near full-length genome characterization of HIV type 1 unique BC recombinant forms from Southern Brazil. AIDS Res. Hum. Retrovir. 2009, 25, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.E.; de Medeiros, R.M.; Junqueira, D.M.; Graf, T.; Passaes, C.P.; Bello, G.; Morgado, M.G.; Guimarães, M.L. Temporal dynamics of HIV-1 circulating subtypes in distinct exposure categories in southern Brazil. Virol. J. 2012, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.P.; Queiroz, B.B.; Stefani, M.M. HIV-1 pol phylogenetic diversity and antiretroviral resistance mutations in treatment naive patients from Central West Brazil. J. Clin. Virol. 2009, 46, 134–139. [Google Scholar] [CrossRef] [PubMed]
- De Medeiros, R.M.; Junqueira, D.M.; Matte, M.C.; Barcellos, N.T.; Chies, J.A.; Matos Almeida, S.E. Co-circulation HIV-1 subtypes B, C, and CRF31_BC in a drug-naive population from Southernmost Brazil: Analysis of primary resistance mutations. J. Med. Virol. 2011, 83, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Graf, T.; Pinto, A.R. The increasing prevalence of HIV-1 subtype C in Southern Brazil and its dispersion through the continent. Virology 2013, 435, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Sanabani, S.S.; Pessoa, R.; Soares de Oliveira, A.C.; Martinez, V.P.; Giret, M.T.; de Menezes Succi, R.C.; Carvalho, K.; Tomiyama, C.S.; Nixon, D.F.; Sabino, E.C.; et al. Variability of HIV-1 genomes among children and adolescents from Sao Paulo, Brazil. PLoS ONE 2013, 8, e62552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, L.F.; Ishak, M.O.; Vallinoto, A.C.; Lemos, J.A.; Azevedo, V.N.; Moreira, M.R.; Souza, M.I.; Fernandes, L.M.; Souza, L.L.; Ishak, R. Molecular epidemiology of HIV type 1 in northern Brazil: Identification of subtypes C and D and the introduction of CRF02_AG in the Amazon region of Brazil. AIDS Res. Hum. Retrovir. 2009, 25, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, R.; Loureiro, P.; Esther Lopes, M.; Carneiro-Proietti, A.B.; Sabino, E.C.; Busch, M.P.; Sanabani, S.S. Ultra-Deep Sequencing of HIV-1 near Full-Length and Partial Proviral Genomes Reveals High Genetic Diversity among Brazilian Blood Donors. PLoS ONE 2016, 11, e0152499. [Google Scholar] [CrossRef] [PubMed]
- Velasco-de-Castro, C.A.; Grinsztejn, B.; Veloso, V.G.; Bastos, F.I.; Pilotto, J.H.; Fernandes, N.; Morgado, M.G. HIV-1 diversity and drug resistance mutations among people seeking HIV diagnosis in voluntary counseling and testing sites in Rio de Janeiro, Brazil. PLoS ONE 2014, 9, e87622. [Google Scholar] [CrossRef] [PubMed]
- Noe, A.; Plum, J.; Verhofstede, C. The latent HIV-1 reservoir in patients undergoing HAART: An archive of pre-HAART drug resistance. J. Antimicrob. Chemother. 2005, 55, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Bon, I.; Alessandrini, F.; Borderi, M.; Gorini, R.; Re, M.C. Analysis of HIV-1 drug-resistant variants in plasma and peripheral blood mononuclear cells from untreated individuals: Implications for clinical management. New Microbiol. 2007, 30, 313–317. [Google Scholar] [PubMed]
- Wirden, M.; Soulie, C.; Valantin, M.A.; Fourati, S.; Simon, A.; Lambert-Niclot, S.; Bonmarchand, M.; Clavel-Osorio, C.; Marcelin, A.G.; Katlama, C.; et al. Historical HIV-RNA resistance test results are more informative than proviral DNA genotyping in cases of suppressed or residual viraemia. J. Antimicrob. Chemother. 2011, 66, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.R.; Tolstrup, M.; Sogaard, O.S.; Jorgensen, L.B.; Gorry, P.R.; Laursen, A.; Ostergaard, L. Transmission of HIV-1 drug-resistant variants: Prevalence and effect on treatment outcome. Clin. Infect. Dis. 2010, 50, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Alencar, C.S.; Nishiya, A.S.; Ferreira, S.; Giret, M.T.; Diaz, R.S.; Sabino, E.C. Evaluation of primary resistance to HIV entry inhibitors among brazilian patients failing reverse transcriptase/protease inhibitors treatment reveal high prevalence of maraviroc resistance-related mutations. AIDS Res. Hum. Retrovir. 2010, 26, 1267–1271. [Google Scholar] [CrossRef] [PubMed]
- Araujo, L.A.; Junqueira, D.M.; de Medeiros, R.M.; Matte, M.C.; Almeida, S.E. Naturally occurring resistance mutations to HIV-1 entry inhibitors in subtypes B, C, and CRF31_BC. J. Clin. Virol. 2012, 54, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Rodes, B.; Labernardiere, J.L.; Benito, J.M.; Toro, C.; Gonzalez-Lahoz, J.; Faudon, J.L.; Clavel, F.; Schapiro, J.; Soriano, V. Evolution of genotypic and phenotypic resistance to Enfuvirtide in HIV-infected patients experiencing prolonged virologic failure. J. Med. Virol. 2004, 74, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E.; Rodes, B.; Lebel-Binay, S.; Faudon, J.L.; Jimenez, V.; Soriano, V. Dynamics of enfuvirtide resistance in HIV-infected patients during and after long-term enfuvirtide salvage therapy. J. Clin. Virol. 2005, 34, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Brindeiro, R.M.; Diaz, R.S.; Sabino, E.C.; Morgado, M.G.; Pires, I.L.; Brigido, L.; Dantas, M.C.; Barreira, D.; Teixeira, P.R.; Tanuri, A. Brazilian Network for Drug Resistance, S. Brazilian Network for HIV Drug Resistance Surveillance (HIV-BResNet): A survey of chronically infected individuals. AIDS 2003, 17, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Gonsalez, C.R.; Alcalde, R.; Nishiya, A.; Barreto, C.C.; Silva, F.E.; de Almeida, A.; Mendonca, M.; Ferreira, F.; Fernandes, S.S.; Casseb, J.; et al. Drug resistance among chronic HIV-1-infected patients naive for use of anti-retroviral therapy in Sao Paulo city. Virus Res. 2007, 129, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.S.; Cardoso, L.P.; Stefani, M.M. Moderate prevalence of transmitted drug resistance and high HIV-1 genetic diversity in patients from Mato Grosso State, Central Western Brazil. J. Med. Virol. 2011, 83, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Inocencio, L.A.; Pereira, A.A.; Sucupira, M.C.; Fernandez, J.C.; Jorge, C.P.; Souza, D.F.; Fink, H.T.; Diaz, R.S.; Becker, I.M.; Suffert, T.A.; et al. Brazilian Network for HIV Drug Resistance Surveillance: A survey of individuals recently diagnosed with HIV. J. Int. AIDS Soc. 2009, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilotto, J.H.; Grinsztejn, B.; Veloso, V.G.; Velasque, L.S.; Friedman, R.K.; Moreira, R.I.; Rodrigues-Pedro, A.; Oliveira, S.M.; Currier, J.S.; Morgado, M.G. Moderate prevalence of transmitted drug resistance mutations among antiretroviral-naive HIV-infected pregnant women in Rio de Janeiro, Brazil. AIDS Res. Hum. Retrovir. 2013, 29, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Sprinz, E.; Netto, E.M.; Patelli, M.; Lima, J.S.; Furtado, J.J.; da Eira, M.; Zajdenverg, R.; Madruga, J.V.; Lewi, D.S.; Machado, A.A.; et al. Primary antiretroviral drug resistance among HIV type 1-infected individuals in Brazil. AIDS Res. Hum. Retrovir. 2009, 25, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.W.; Harrigan, P.R.; Huang, W.; Bates, M.; Williamson, D.W.; McCune, J.M.; Price, R.W.; Spudich, S.S.; Lampiris, H.; Hoh, R.; et al. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J. Infect. Dis. 2006, 194, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Koot, M.; Keet, I.P.; Vos, A.H.; de Goede, R.E.; Roos, M.T.; Coutinho, R.A.; Miedema, F.; Schellekens, P.T.; Tersmette, M. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 1993, 118, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Moyle, G.J.; Wildfire, A.; Mandalia, S.; Mayer, H.; Goodrich, J.; Whitcomb, J.; Gazzard, B.G. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J. Infect. Dis. 2005, 191, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, G.; Tresoldi, E.; Bjorndal, A.; Fredriksson, R.; Colognesi, C.; Deng, H.K.; Malnati, M.S.; Plebani, A.; Siccardi, A.G.; Littman, D.R.; et al. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 1997, 3, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Schuitemaker, H.; Koot, M.; Kootstra, N.A.; Dercksen, M.W.; de Goede, R.E.; van Steenwijk, R.P.; Lange, J.M.; Schattenkerk, J.K.; Miedema, F.; Tersmette, M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: Progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 1992, 66, 1354–1360. [Google Scholar] [PubMed]
- Skrabal, K.; Low, A.J.; Dong, W.; Sing, T.; Cheung, P.K.; Mammano, F.; Harrigan, P.R. Determining human immunodeficiency virus coreceptor use in a clinical setting: Degree of correlation between two phenotypic assays and a bioinformatic model. J. Clin. Microbiol. 2007, 45, 279–284. [Google Scholar] [CrossRef] [PubMed]
- de Azevedo, S.S.D.; Caetano, D.G.; Cortes, F.H.; Teixeira, S.L.M.; Dos Santos Silva, K.; Hoagland, B.; Grinsztejn, B.; Veloso, V.G.; Morgado, M.G.; Bello, G. Highly divergent patterns of genetic diversity and evolution in proviral quasispecies from HIV controllers. Retrovirology 2017, 14, 29. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Subdomain | Mutation | ARV Class Associated with Resistance |
|---|---|---|
| CN | E312Q | NRTI |
| Y318F/W | NNRTI | |
| G335D/C (polymorphism) | NRTI | |
| N348I | NRTI and NNRTI | |
| A360I/V | NRTI | |
| V365I | NRTI | |
| T369I/V | NRTI and NNRTI | |
| A371V | NRTI | |
| A376S | NRTI and NNRTI | |
| E399D/G | NRTI and NNRTI | |
| A400T (polymorphism) | NRTI | |
| RH | D488E | NRTI |
| Q509L | NRTI and NNRTI | |
| Q547K | NRTI |
| Characteristic | N |
|---|---|
| Males (%) | 24 (75%) |
| Age (years) (mean ± SD) | 40 ± 12.3 |
| Median baseline CD4+ T-cell counts (cells/mm3; IQR50) | 712.5 (606.5–856) |
| Median baseline CD8+ T-cell counts (cells/mm3; IQR50) | 657.5 (529–1047.25) |
| Median time since HIV diagnosis (years; IQR50) | 4.7 (3.9–6.5) |
| Median time from HIV diagnosis to antiretroviral therapy initiation (years; IQR50) * | 1.2 (0.6–2.8) |
| Median time of treatment (years; IQR50) | 3.1 (2.4–3.9) |
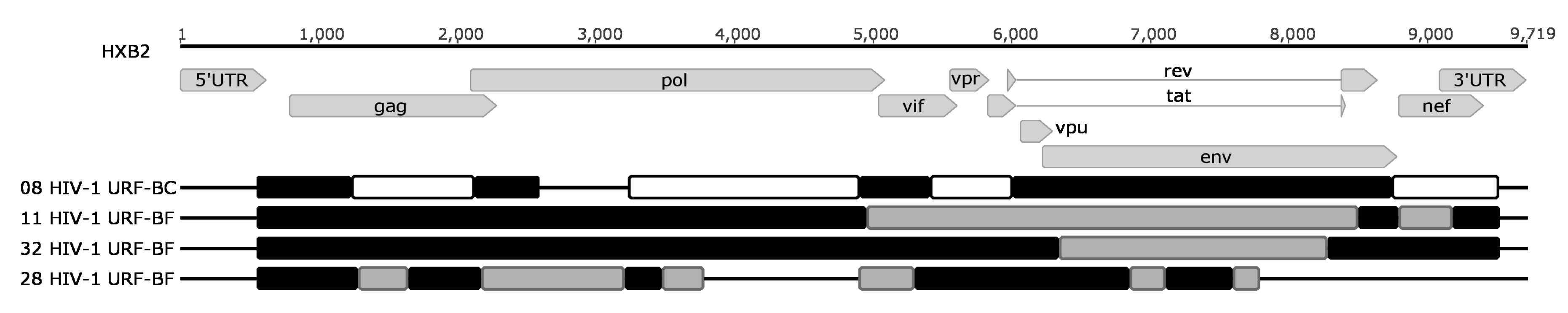
| Patient | Protease Mutations (Coverage; Frequency) | Reverse Transcriptase Mutations (Coverage; Frequency) | RT Connection Mutations (Coverage; Frequency) | RT RNase H Mutation | Integrase Mutations (Coverage; Frequency) | Envelope Mutations (Coverage; Frequency) | HAART Regimen | Tropism 1 | Subtype/URF |
|---|---|---|---|---|---|---|---|---|---|
| 1 | - | M184V (11,979; 2.0%) | - | - | S147G (8945; 1.1%) | - | AZT + 3TC + NVP | 100.0% X4/R5X4 | B |
| 2 # | - | - | - | - | R263K (2201; 6.2%) | - | AZT + 3TC + EFV | 63.0% X4/R5X4 | B |
| 3 | - | - | - | - | - | - | TDF + 3TC + EFV | 97.2% R5 | B |
| 5 | I47V (3589; 10.3%) | - | - | - | - | - | AZT + 3TC + ATV | 100% R5 | B |
| 8 | - | NA | E399D (2701; 99.2%) | - | - | - | TDF + 3TC + EFV | 97.1% R5 | BC |
| 11 | D30N (8012; 2.8%) M46I (8639; 1.9%) | M41L (5444; 99.8%) | T369V (9726; 39.6%) E399G (8334; 4.9%) | - | - | - | TDF + 3TC + EFV | 96.1% R5 | BF |
| 12 | - | - | - | - | - | - | TDF + 3TC + EFV | 84.8% R5 | B |
| 13 | - | E138K (4909; 1.3%) | - | - | - | V38A (2151; 3.0%) | TDF + 3TC + EFV | 99.0% X4/R5X4 | B |
| 14 | - | - | - | NA | R263K * (1776; 11.0%) | - | AZT + 3TC + LPV/r | 97.8% R5 | B |
| 15 | - | - | E399G (14,872; 1.5%) | - | - | - | TDF + 3TC + EFV | 97.7% R5 | B |
| 16 | D30N (4574; 49.3%) M46I (4378; 45.6%) | - | - | - | - | - | AZT + 3TC + FPV/r | NA | B |
| 18 | - | A62V (4116; 1.0%) | - | - | - | NA | TDF + 3TC + EFV | 91.6% R5 | B |
| 19 | - | NA | A376S (6663; 99.9%) | - | - | - | AZT + 3TC + EFV | 99.6% R5 | B |
| 20 | - | - | - | NA | NA | NA | TDF + 3TC + EFV | 100.0% R5 | B |
| 21 | - | L210W (12,583; 100.0%) | - | - | - | - | TDF + EFV + FTC | 99.3% R5 | B |
| 22 | - | NA | A376S * (1082; 94.9%) | - | - | - | TDF + 3TC + EFV | 100.0% R5 | B |
| 23 | - | - | - | - | T97A (4113; 2.1%) | - | TDF + 3TC + EFV | 96.0% R5 | B |
| 26 | - | NA | NA | NA | NA | - | TDF + 3TC + EFV | 100.0% X4/R5X4 | B |
| 27 | - | NA | - | - | - | - | TDF + 3TC + EFV | 99.9% R5 | B |
| 28 | - | M41L (5791; 99.5%) D67N (6465; 72.9%) K70R (6475; 77.2%) T215Y (9219; 38.7%) | E399D (2051; 99.9%) | NA | NA | - | TDF + 3TC + EFV | 99.1% R5 | BF |
| 29 | - | - | E399D (5209; 100.0%) | - | T66I (7444; 31.0%) | NA | TDF + 3TC + EFV | 99.7% X4/R5X4 | B |
| 31 | - | M184V (2708; 1.3%) | - | - | - | - | TDF + 3TC + EFV | 100.0% R5 | B |
| 32 | - | V179D (7146; 99.3%) M184V (7127; 8.0%) | - | - | - | - | TDF + 3TC + EFV | 100.0% R5 | BF |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, B.M.; Siqueira, J.D.; Garrido, M.M.; Botelho, O.M.; Prellwitz, I.M.; Ribeiro, S.R.; Soares, E.A.; Soares, M.A. Characterization of HIV-1 Near Full-Length Proviral Genome Quasispecies from Patients with Undetectable Viral Load Undergoing First-Line HAART Therapy. Viruses 2017, 9, 392. https://doi.org/10.3390/v9120392
Alves BM, Siqueira JD, Garrido MM, Botelho OM, Prellwitz IM, Ribeiro SR, Soares EA, Soares MA. Characterization of HIV-1 Near Full-Length Proviral Genome Quasispecies from Patients with Undetectable Viral Load Undergoing First-Line HAART Therapy. Viruses. 2017; 9(12):392. https://doi.org/10.3390/v9120392
Chicago/Turabian StyleAlves, Brunna M., Juliana D. Siqueira, Marianne M. Garrido, Ornella M. Botelho, Isabel M. Prellwitz, Sayonara R. Ribeiro, Esmeralda A. Soares, and Marcelo A. Soares. 2017. "Characterization of HIV-1 Near Full-Length Proviral Genome Quasispecies from Patients with Undetectable Viral Load Undergoing First-Line HAART Therapy" Viruses 9, no. 12: 392. https://doi.org/10.3390/v9120392
APA StyleAlves, B. M., Siqueira, J. D., Garrido, M. M., Botelho, O. M., Prellwitz, I. M., Ribeiro, S. R., Soares, E. A., & Soares, M. A. (2017). Characterization of HIV-1 Near Full-Length Proviral Genome Quasispecies from Patients with Undetectable Viral Load Undergoing First-Line HAART Therapy. Viruses, 9(12), 392. https://doi.org/10.3390/v9120392