Deep Sequencing Analysis of RNAs from Citrus Plants Grown in a Citrus Sudden Death-Affected Area Reveals Diverse Known and Putative Novel Viruses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Extraction and Sequencing
2.3. RNA-Seq and Small RNA Bioinformatics Analysis
2.4. Validation of Candidate Viruses
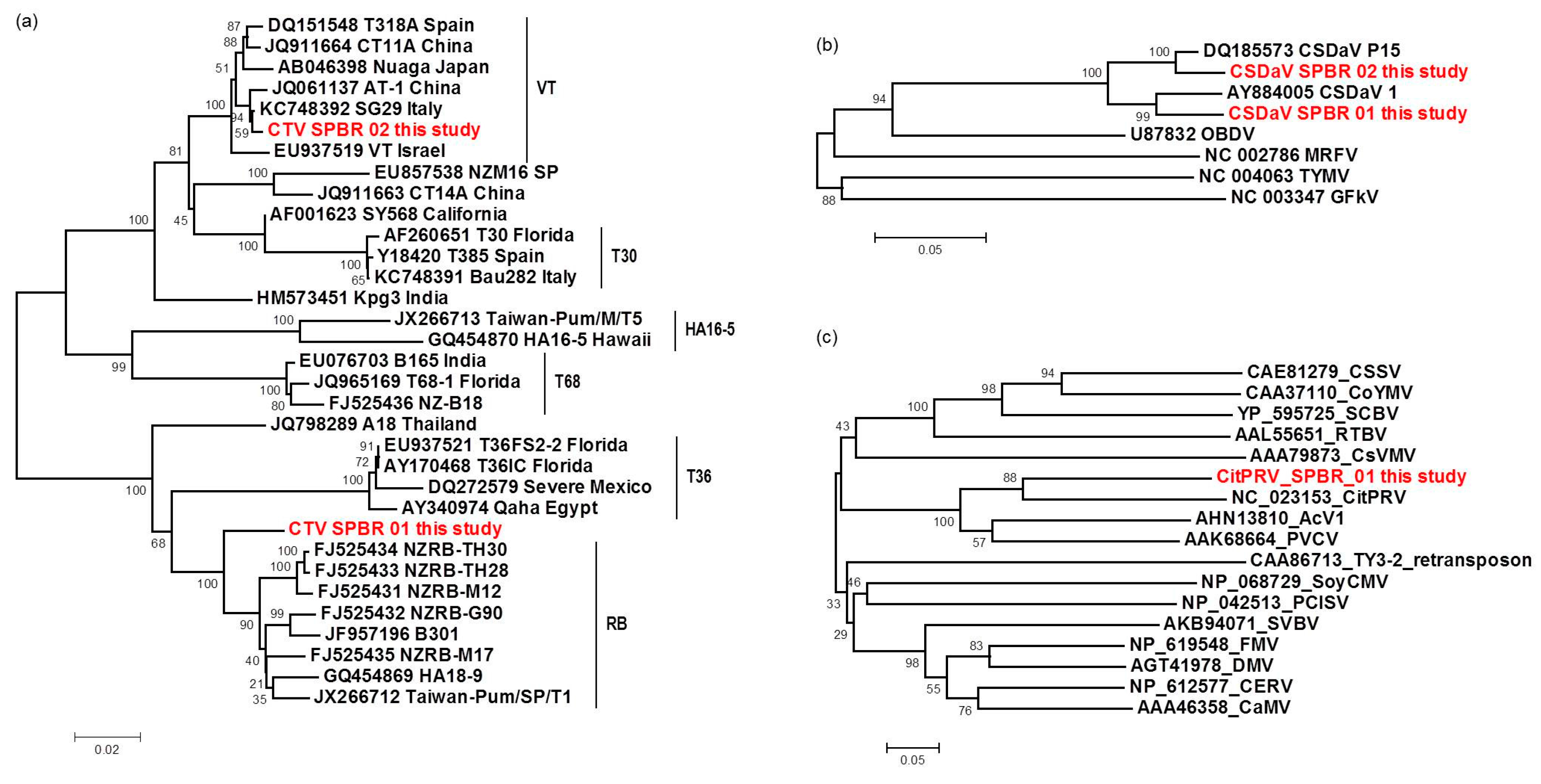
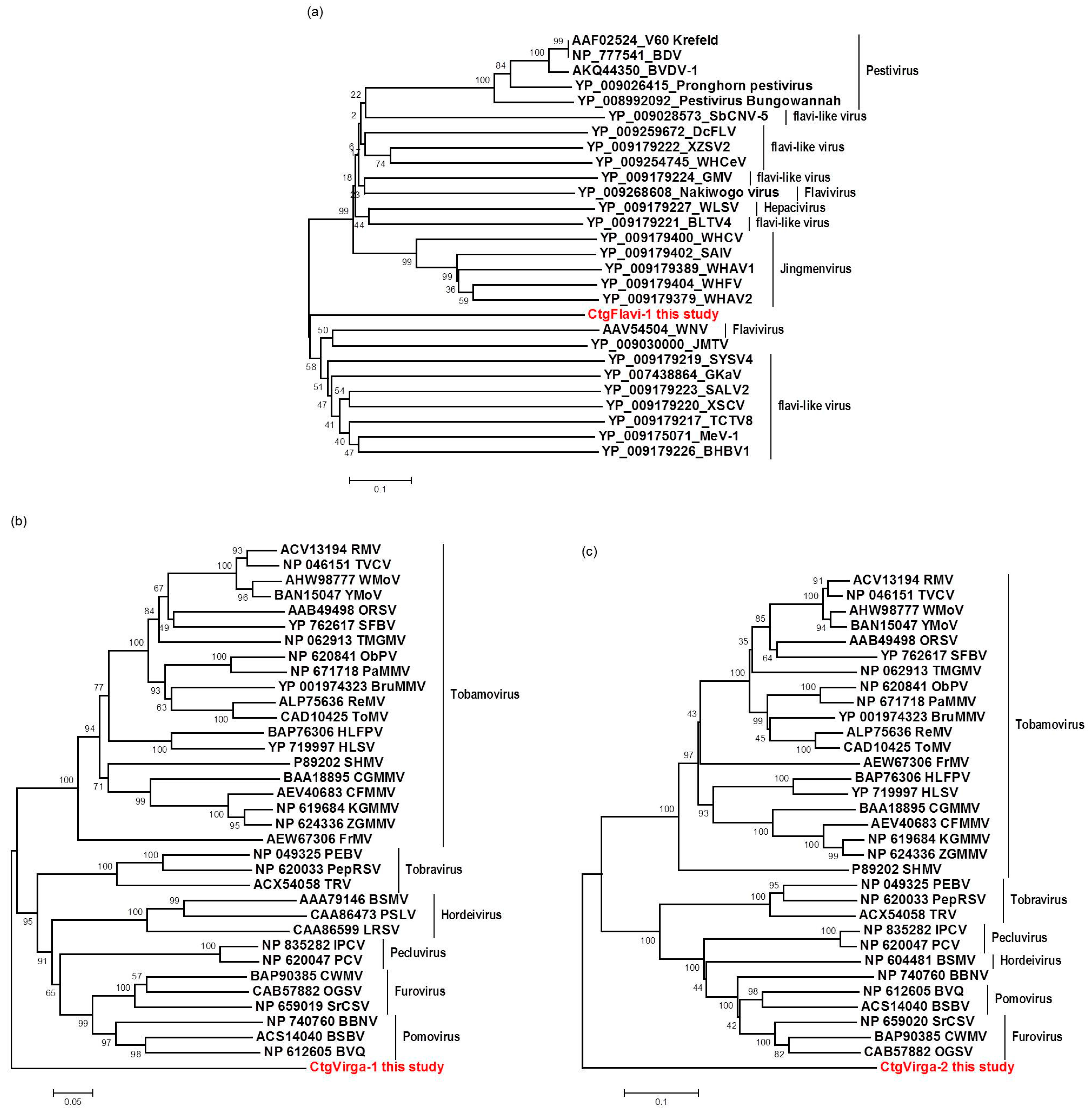
2.5. Phylogenetic Analysis
3. Results
3.1. General Analysis of the RNA-Seq and Small RNA Libraries
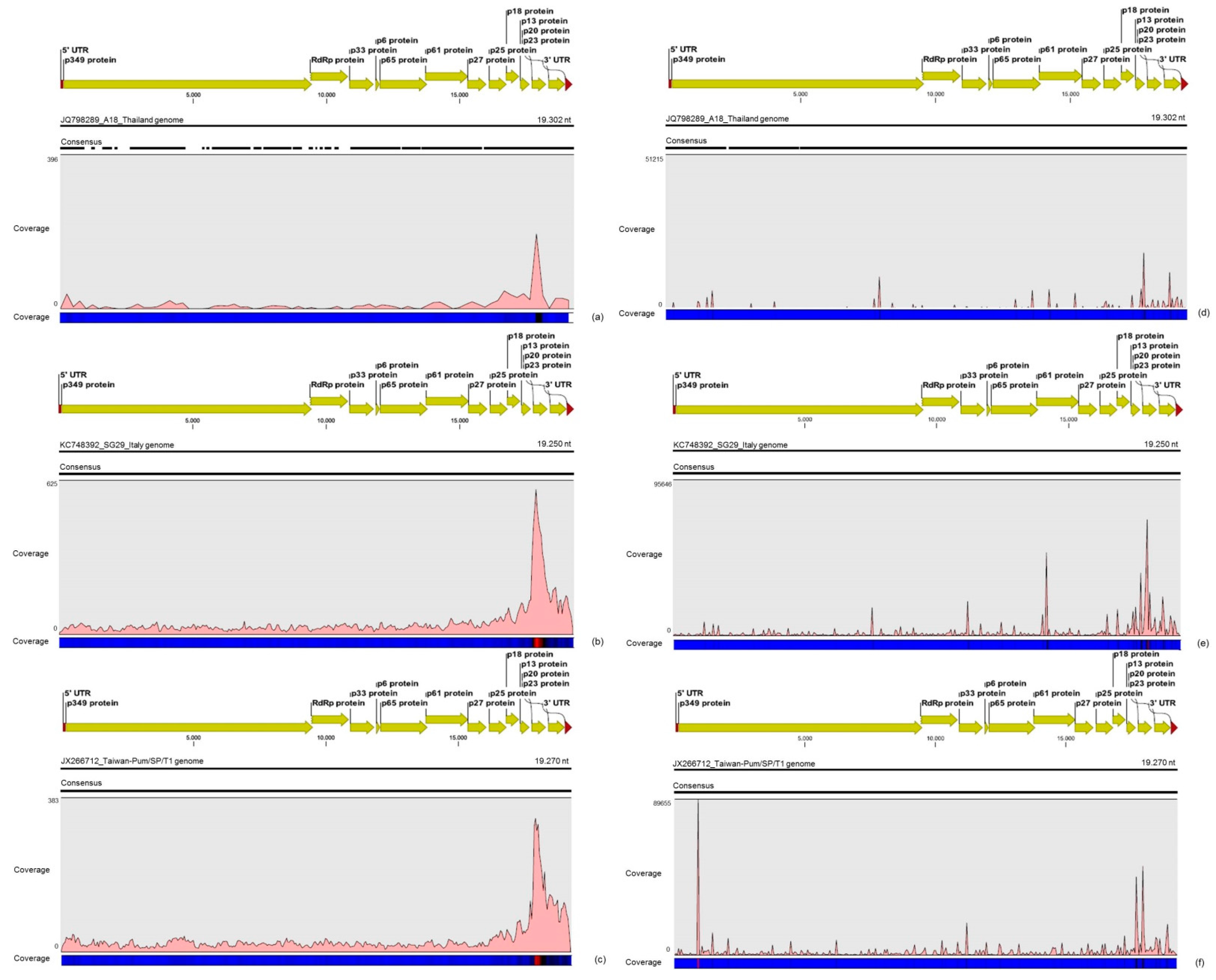
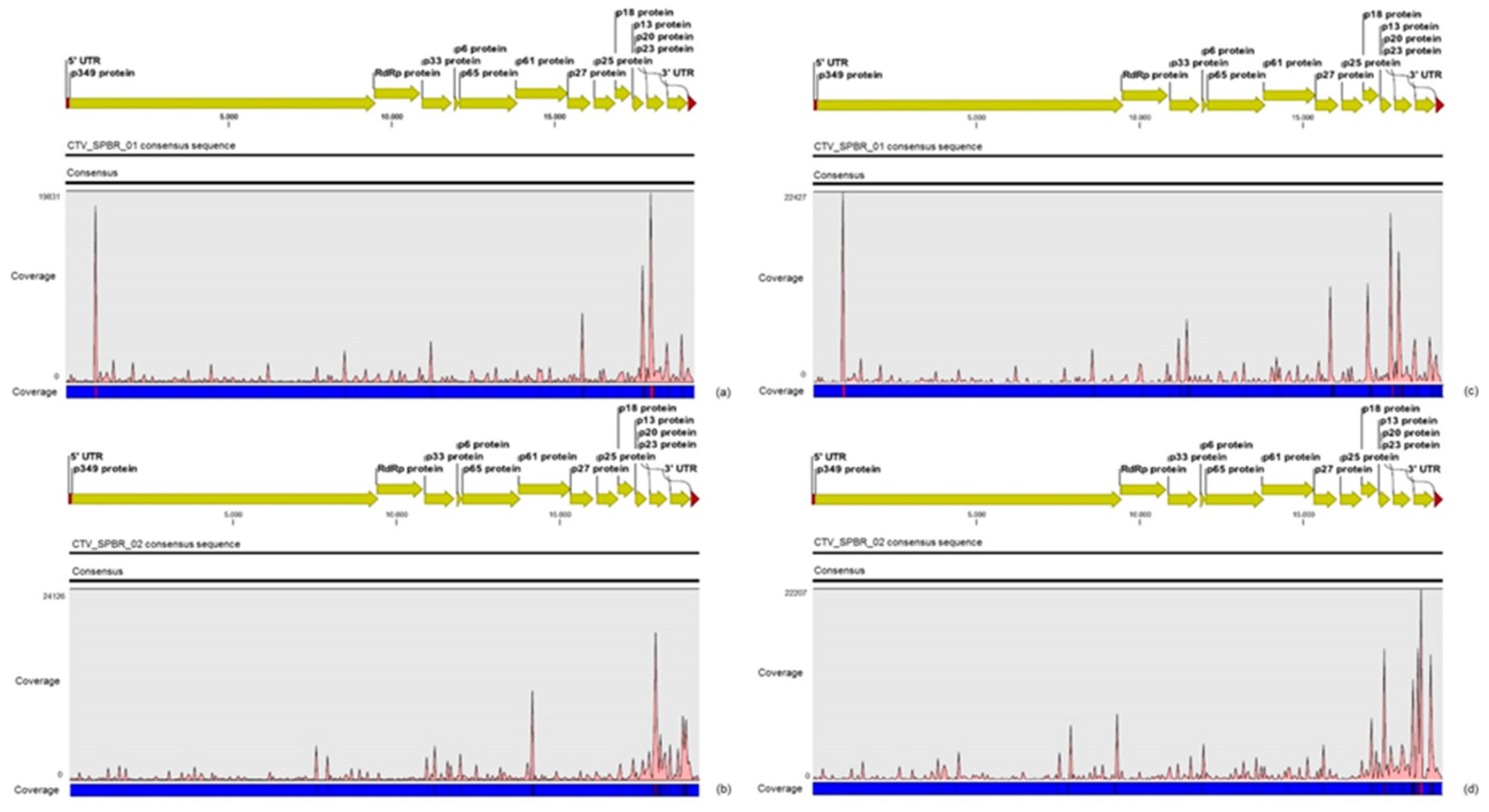
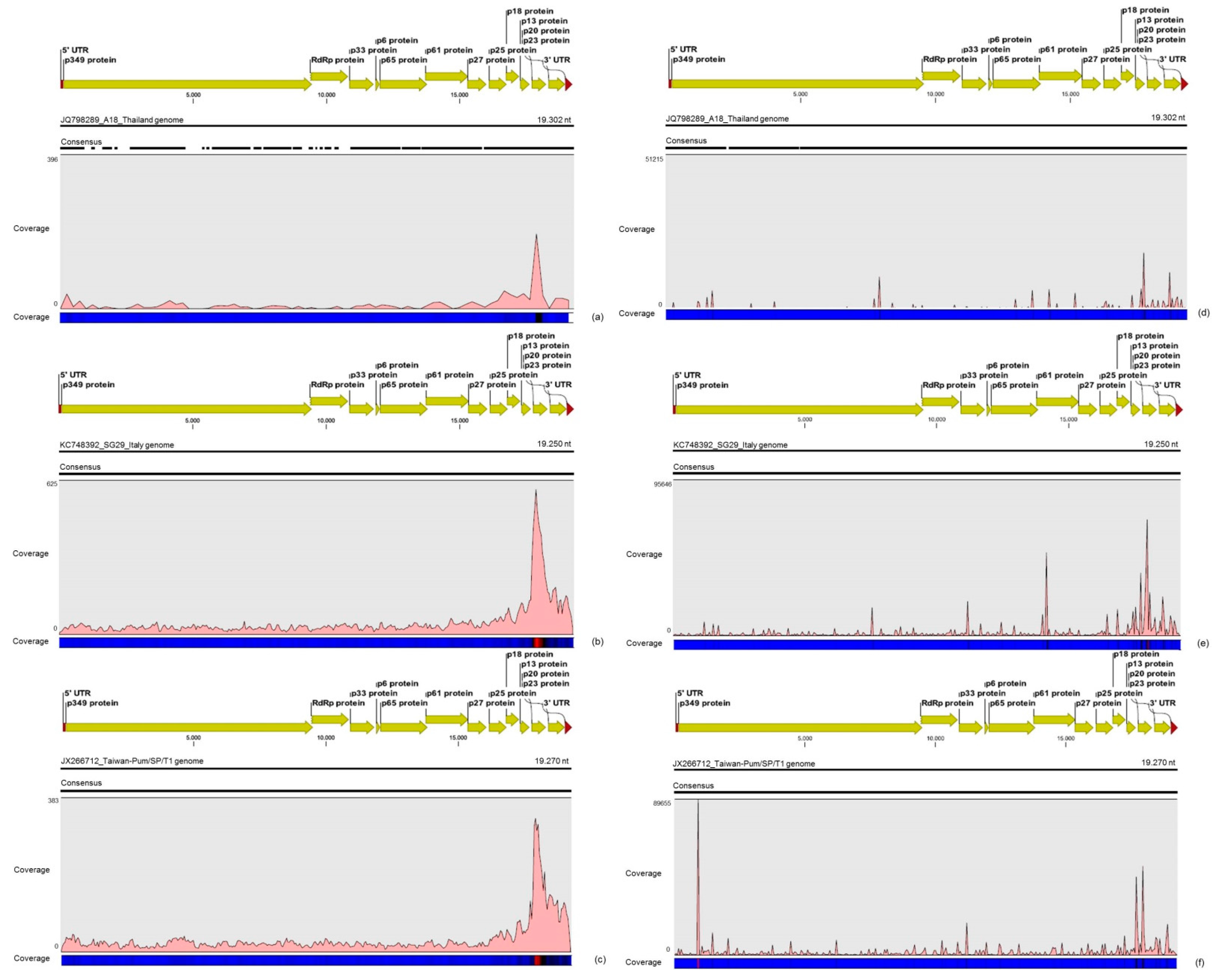
3.2. Contigs Derived from Citrus Tristeza Virus
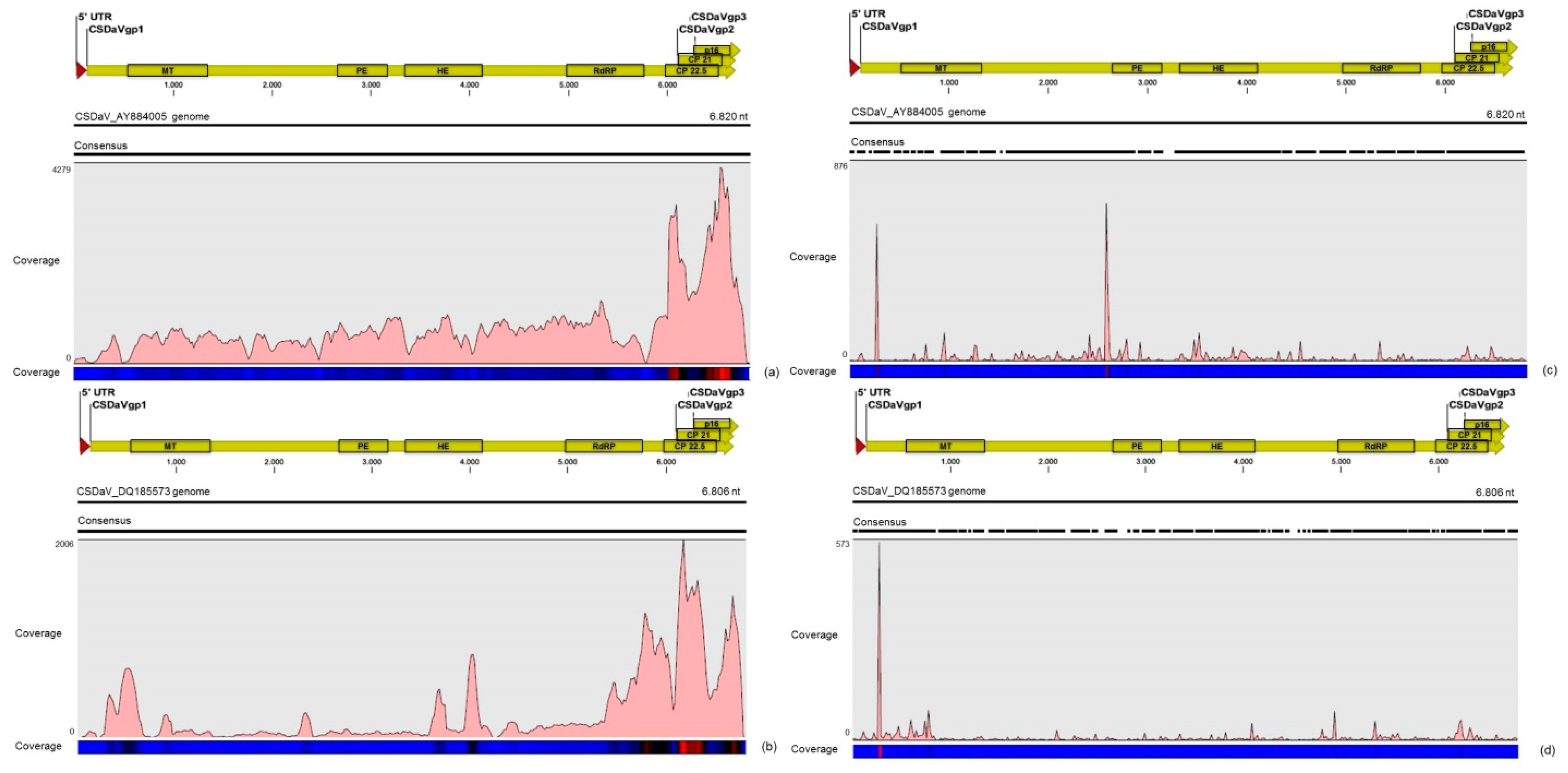
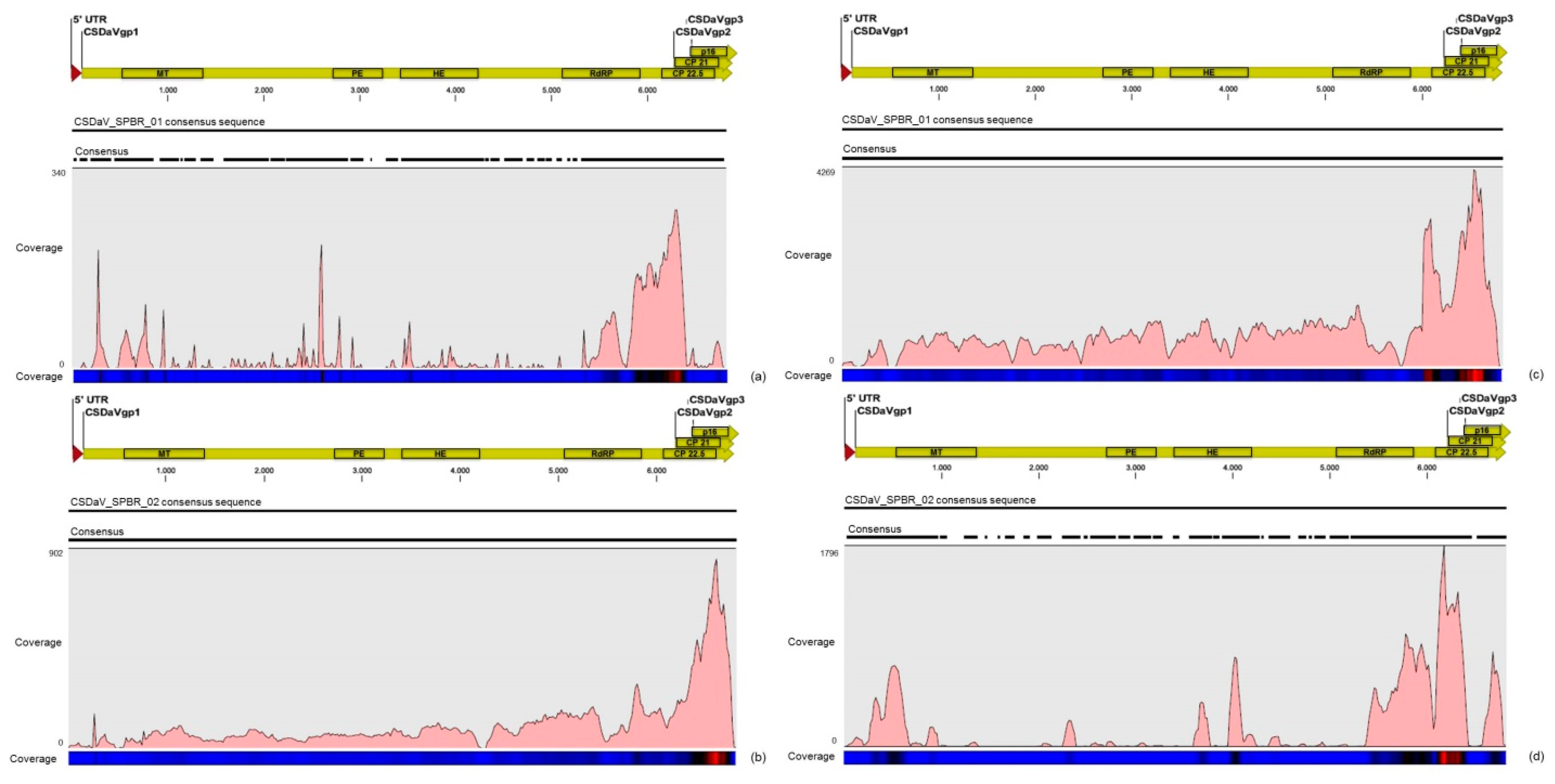
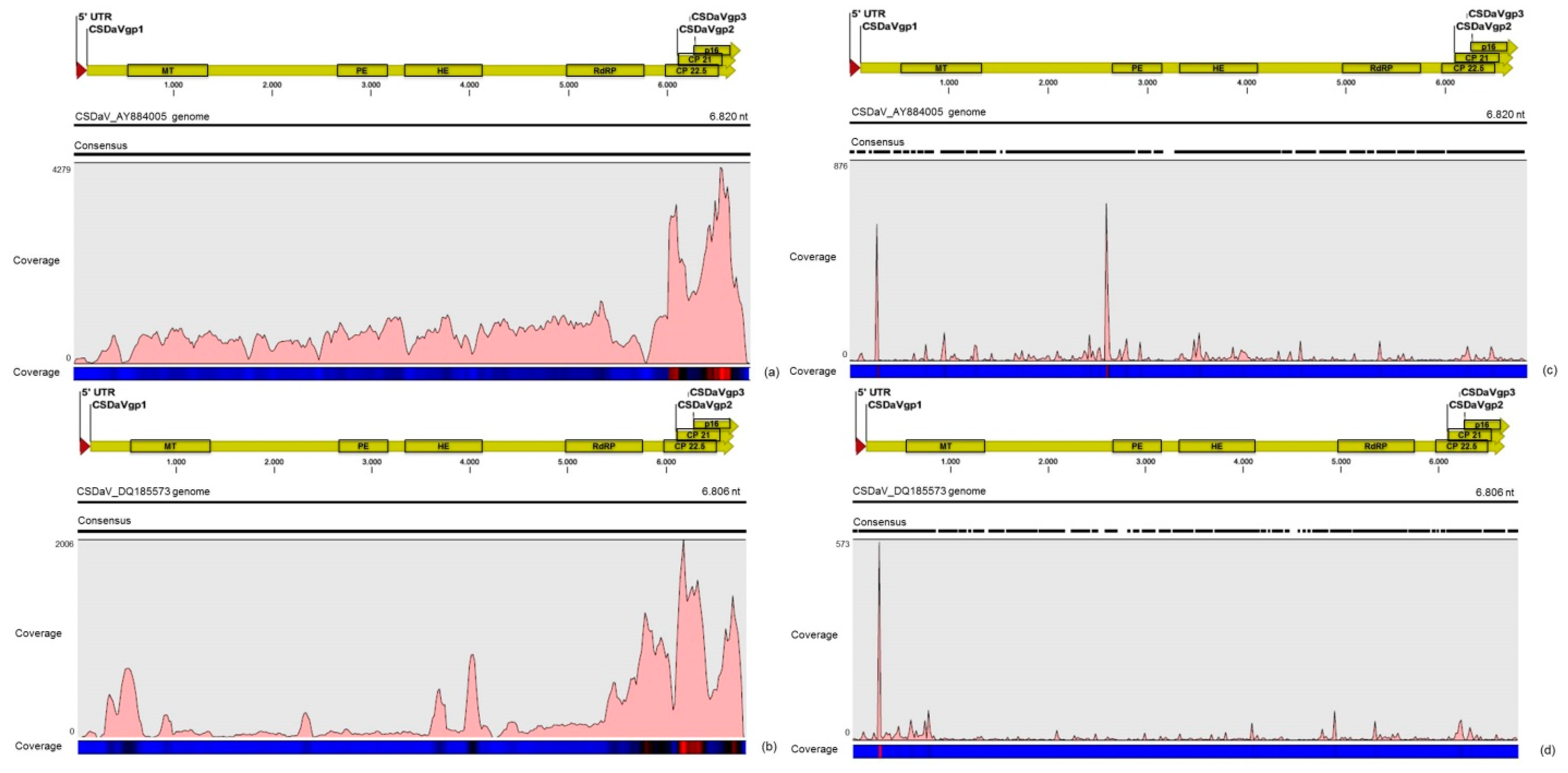
3.3. Contigs Derived from Citrus Sudden Death-Associated Virus
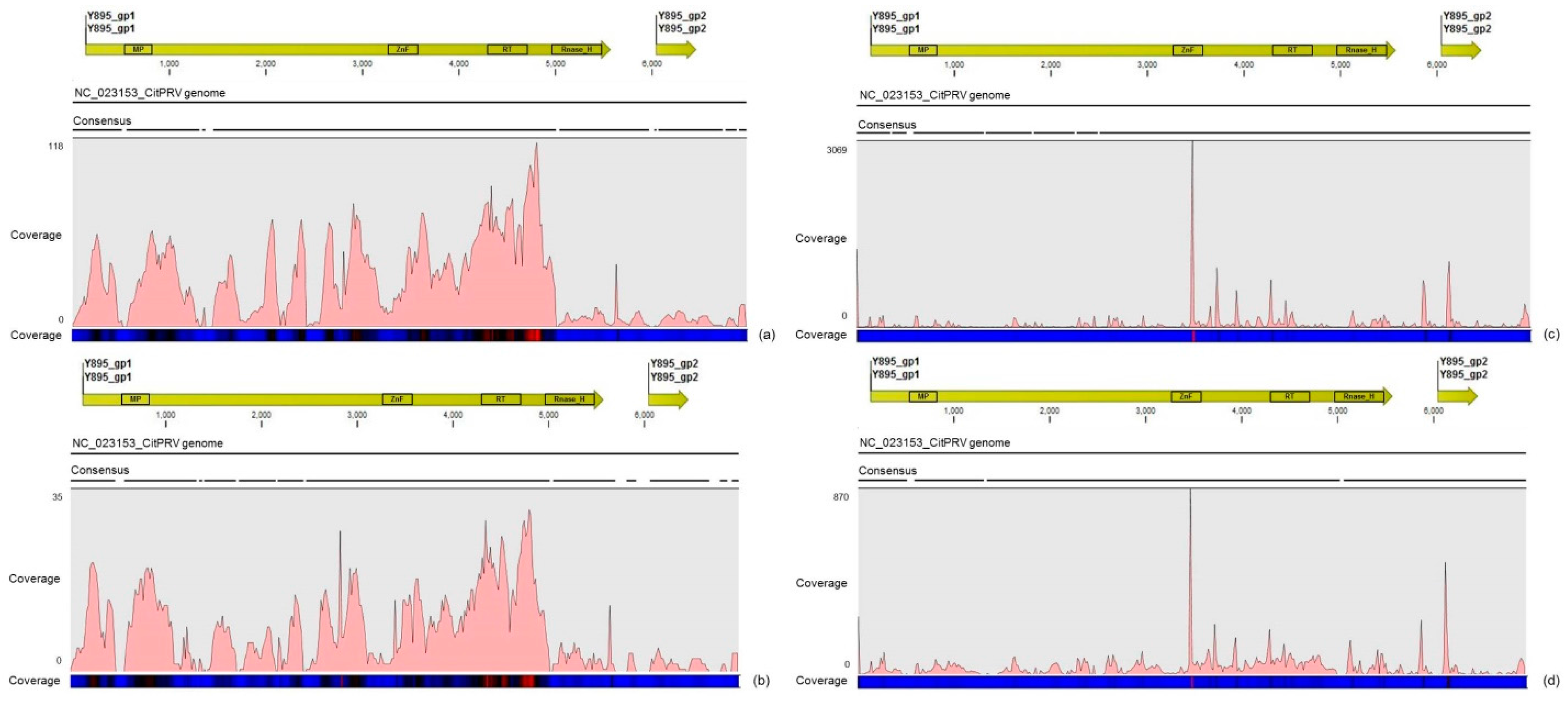
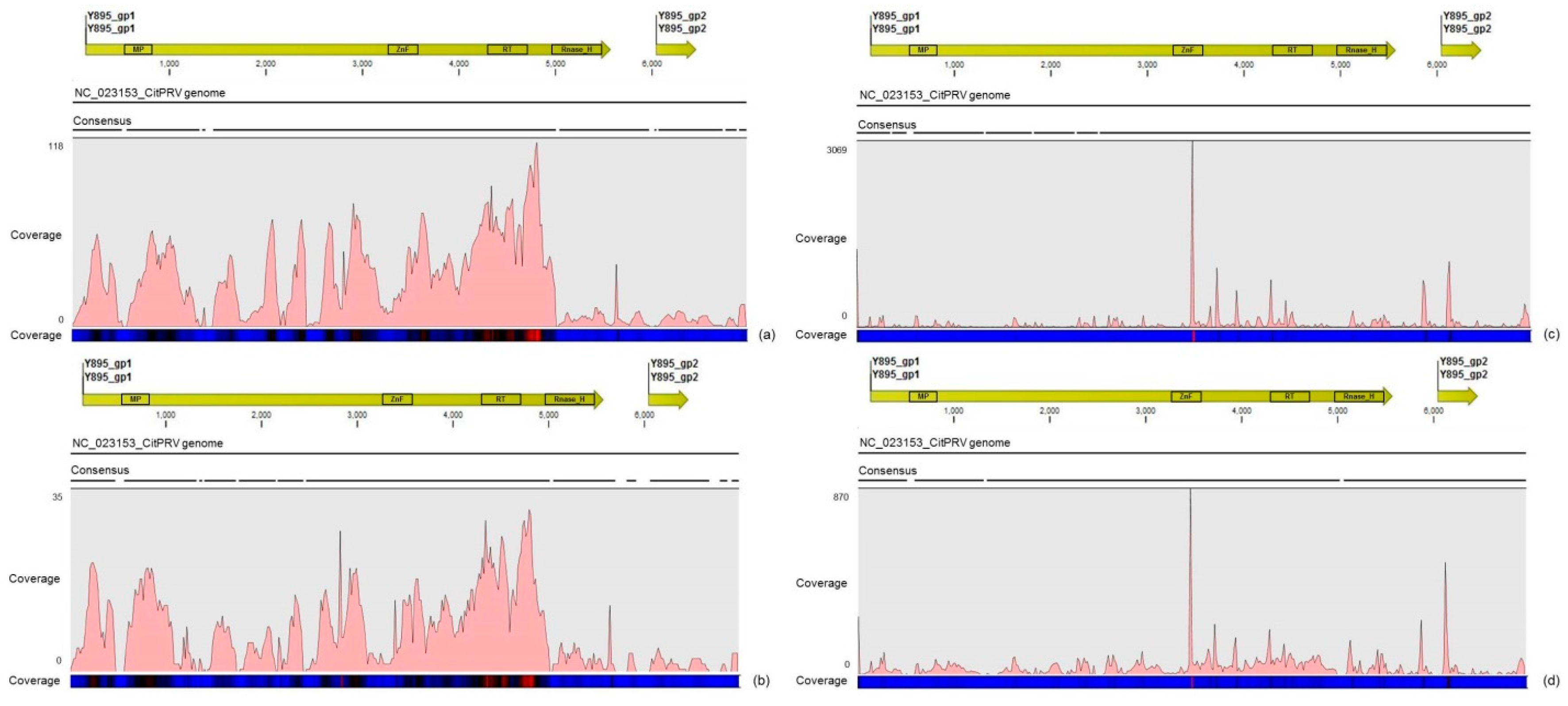
3.4. Description of the Distinct Viral Sequences Detected in the RNA-Seq Libraries
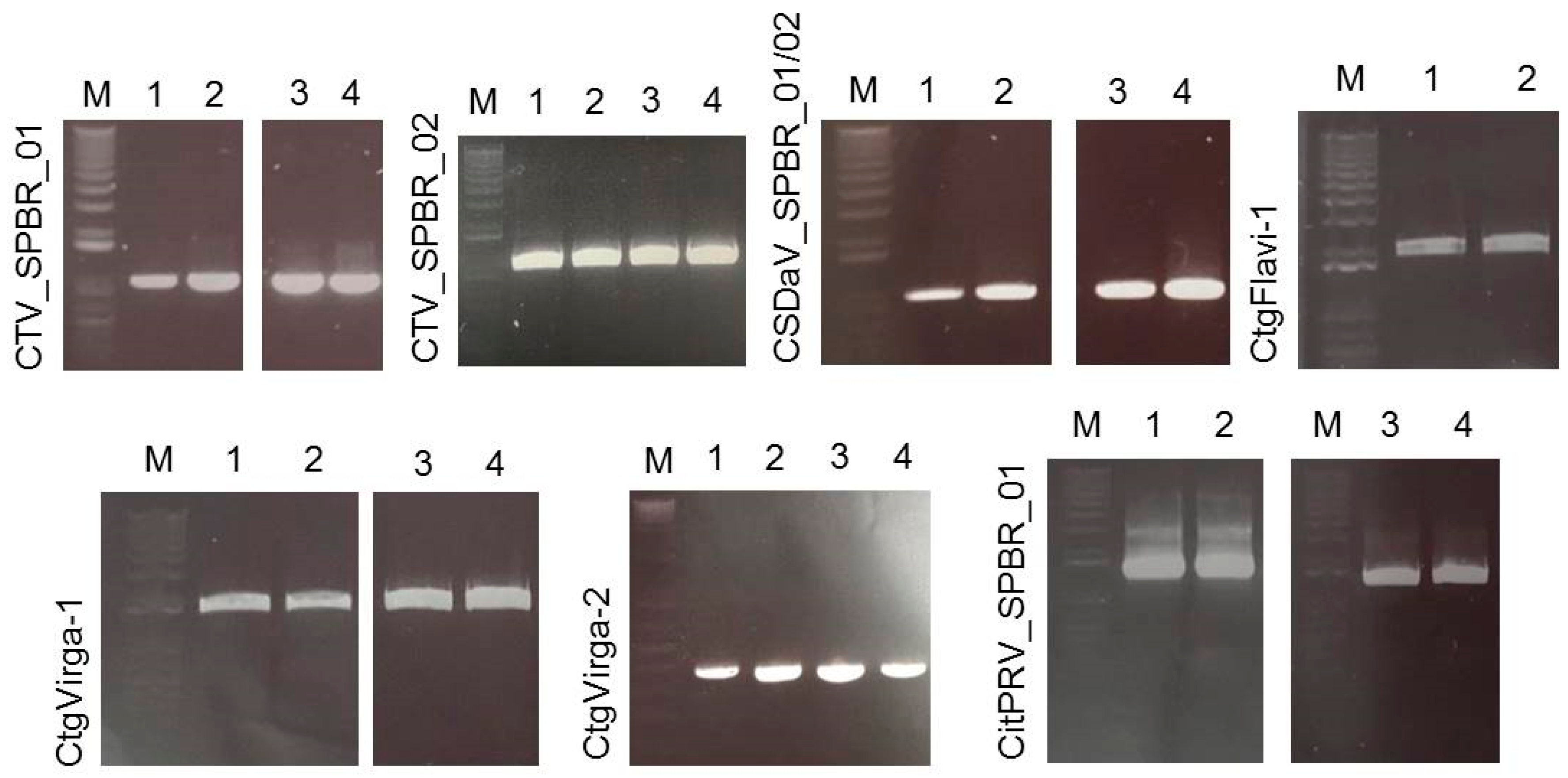
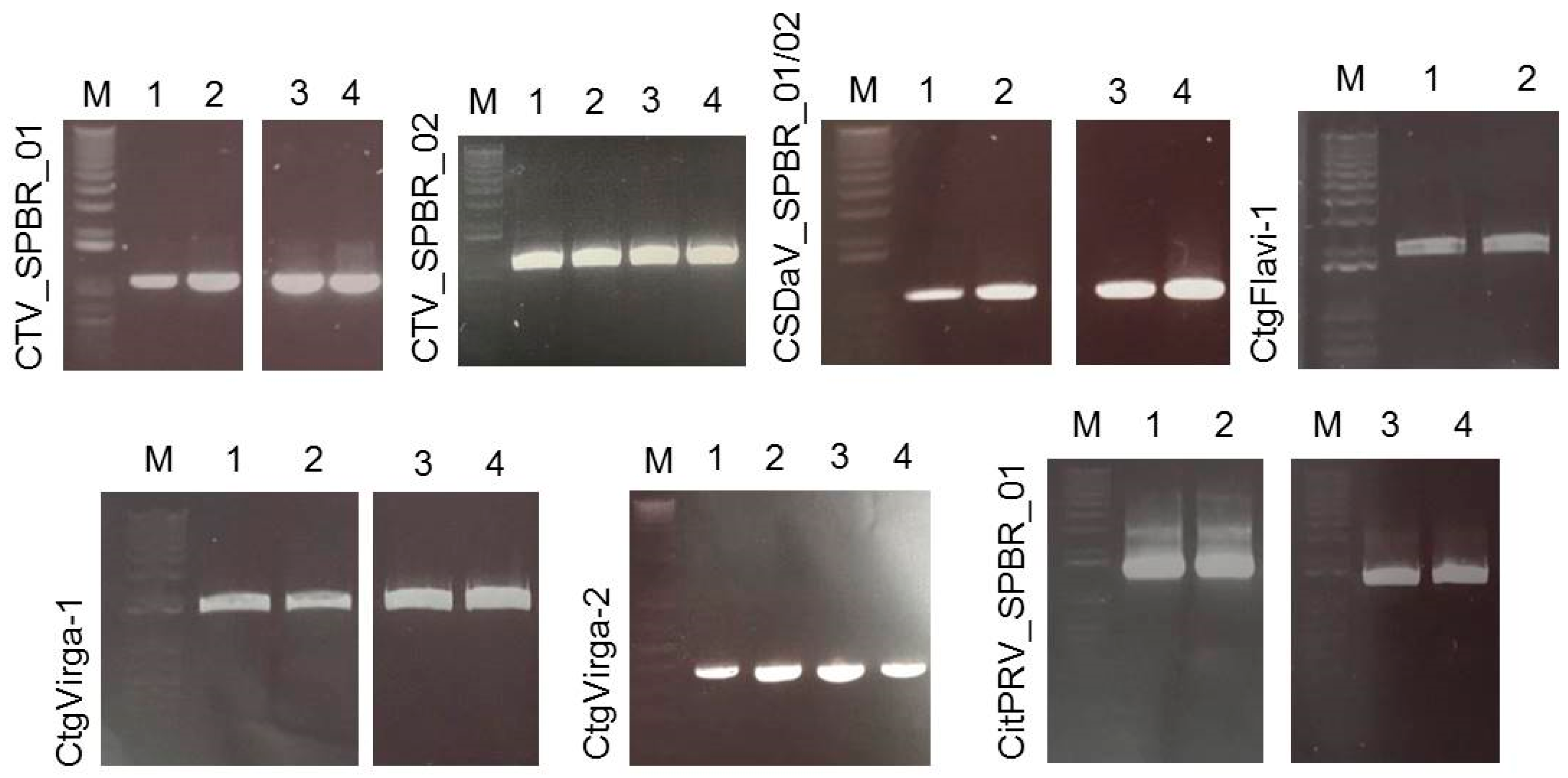
3.5. Validation of Viral Sequences by RT-PCR and Sanger Sequencing
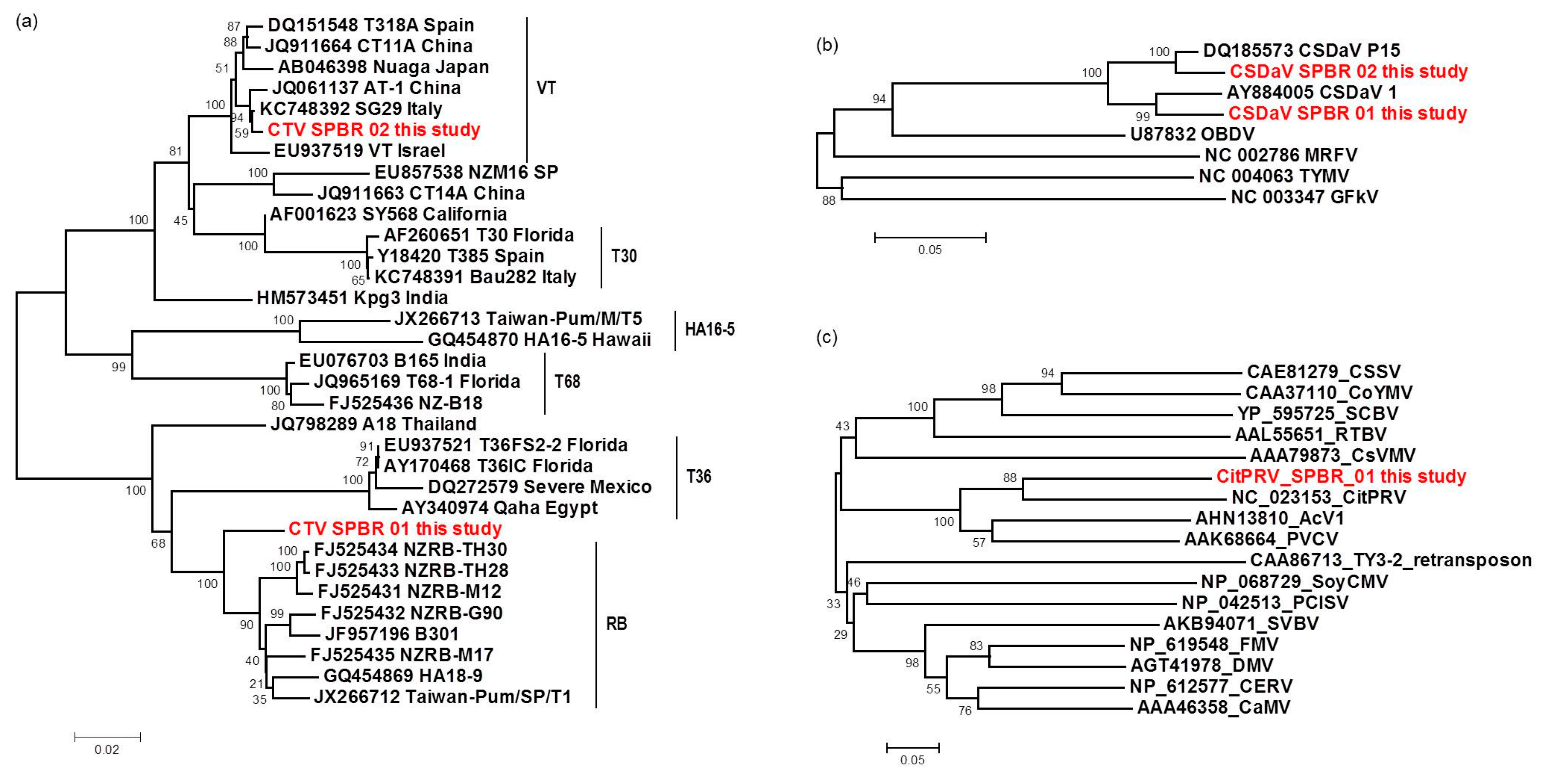
3.6. Sequence and Phylogenetic Analysis of the Viral Sequences Related to the CTV, CSDaV and CitPRV, the Known Viruses Detected in This Study
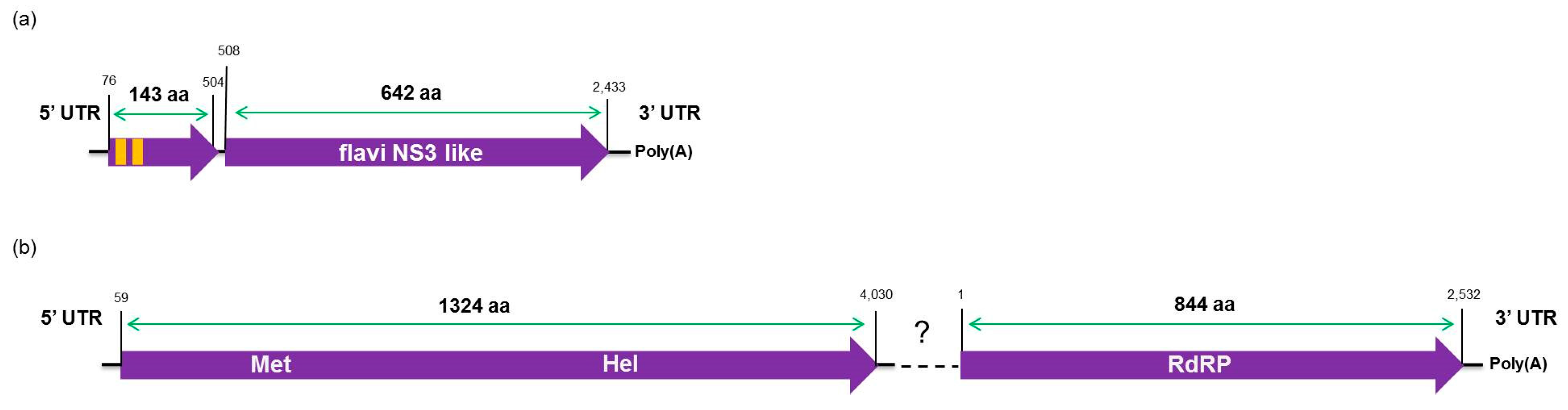
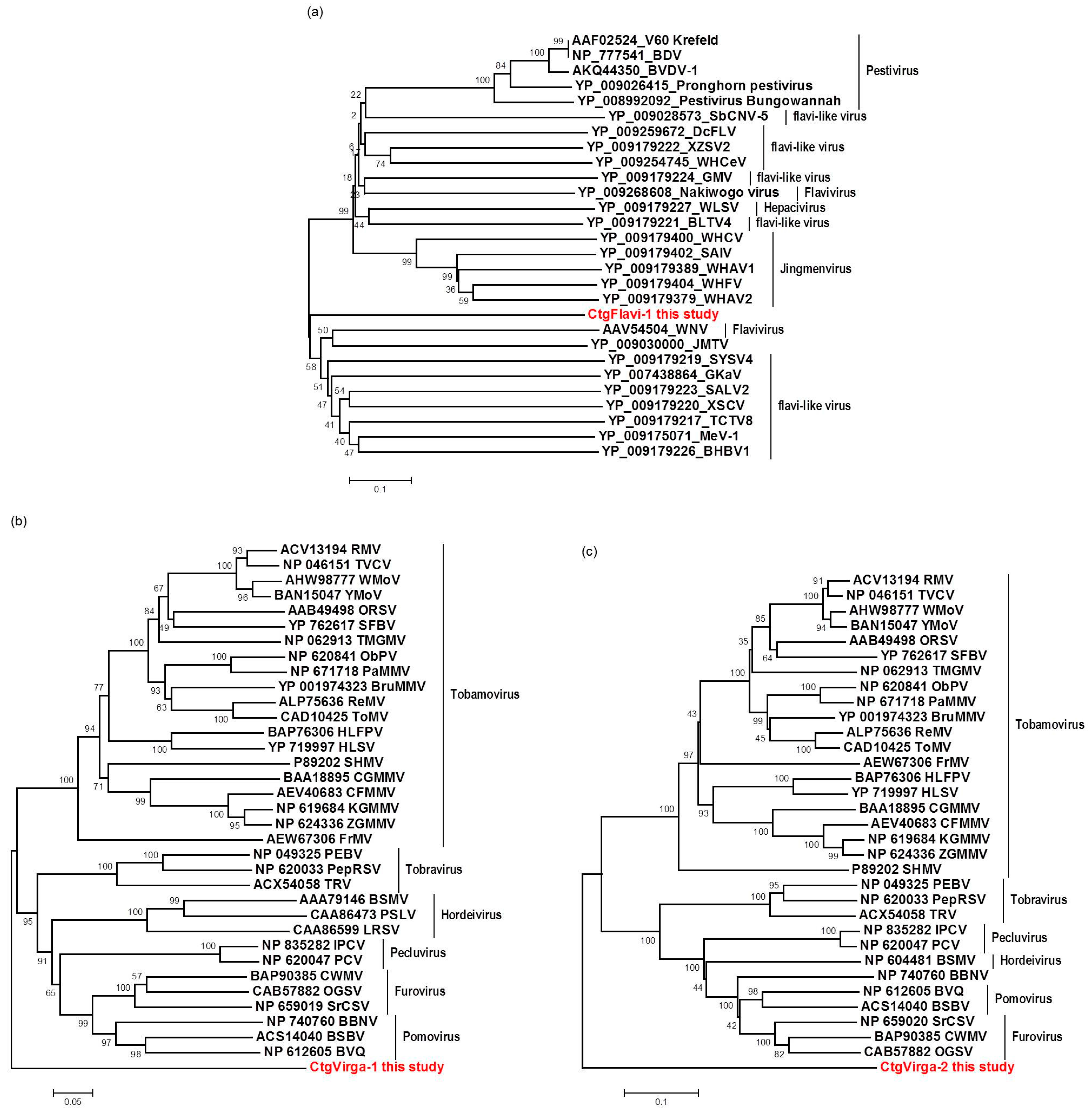
3.7. Phylogenetic Analysis and Preliminary Genome Characterization of the Unknown Viral Sequences Identified in This Study
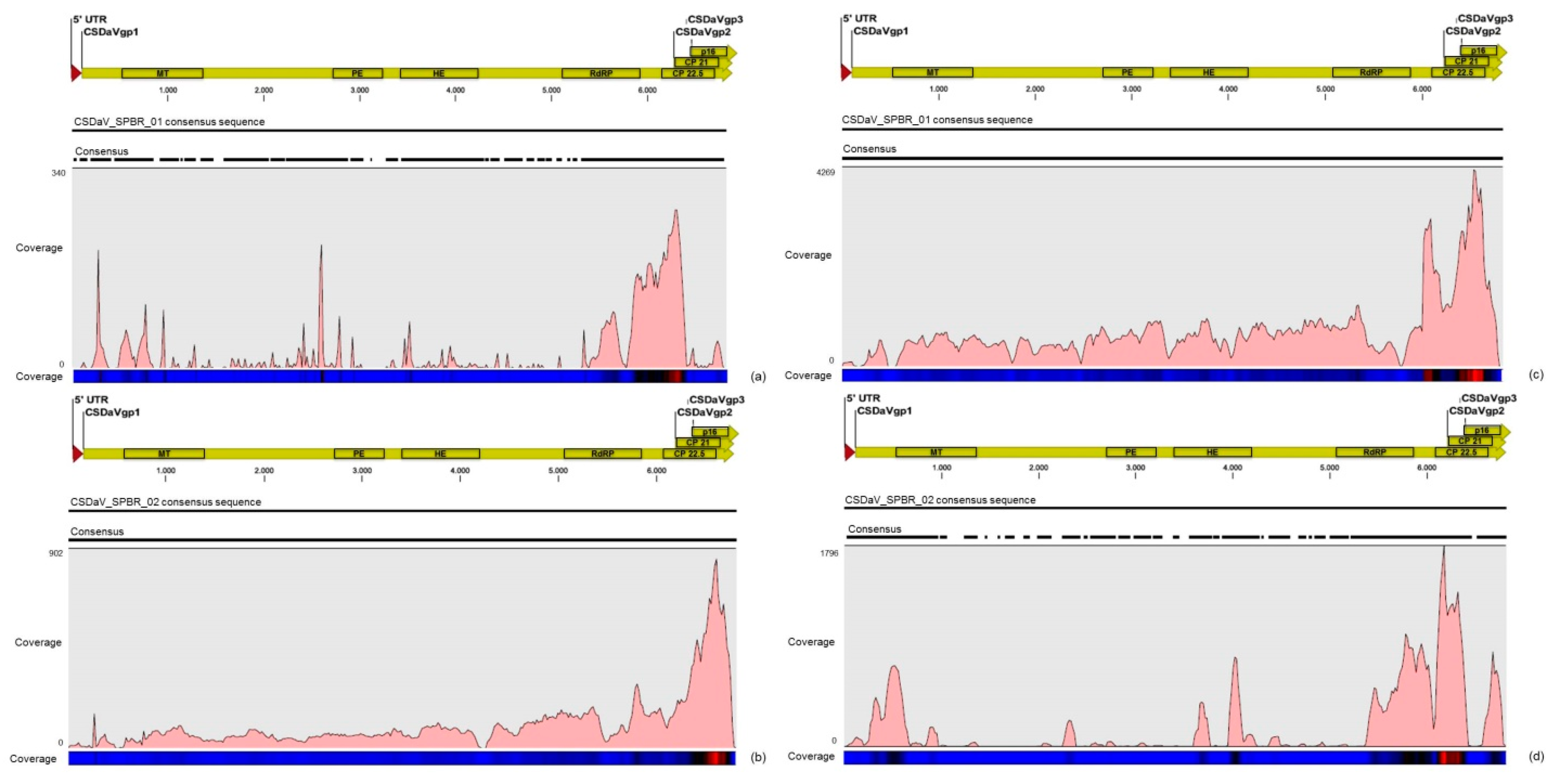
3.8. Comparison of Viral Sequences Derived from CSD-Symptomatic and Asymptomatic Plants
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Müller, G.W.; de Negri, J.D.; Aguilar-Vildoso, C.I.; Mattos, D., Jr.; Pompeu, J., Jr.; Teófilo Sobrinho, J.; Machado, M.A. Citrus sudden death: A new citrus disease in Brazil. In Proceedings of the XV Conference of the International Organization of Citrus Virologists, Paphos, Chipre; 2001; p. 100. [Google Scholar]
- Gomes, C.P.C.; Nagata, T.; de Jesus, W.C., Jr.; Borges Neto, C.R.; Pappas, G.J., Jr.; Martin, D.P. Genetic variation and recombination of RdRp and HSP 70h genes of Citrus tristeza virus isolates from orange trees showing symptoms of citrus sudden death disease. Virol. J. 2008, 5. [Google Scholar] [CrossRef] [PubMed]
- Román, M.P.; Cambra, M.; Juarez, J.; Moreno, P.; Duran-Vila, N.; Tanaka, F.A.O.; Alves, E.; Kitajima, E.W.; Yamamoto, P.T.; Bassanezi, R.B.; et al. Sudden death of citrus in Brazil: A graft-transmissible bud union disease. Plant Dis. 2004, 88, 453–467. [Google Scholar] [CrossRef]
- Bassanezi, R.B.; Montesino, L.H.; Sanches, A.L.; Spósito, M.B.; Stuchi, E.S.; Barbosa, J.C. Effect of citrus sudden death on yield and quality of sweet orange cultivars in Brazil. Plant Dis. 2007, 91, 1407–1412. [Google Scholar] [CrossRef]
- Coletta Filho, H.D.; Centro de Citricultura Sylvio Moreira, Instituto Agronômico, Cordeiropolis, S.P. Brazil. Personal communication, 2014.
- Bassanezi, R.B.; Bergamin Filho, A.; Amorim, L.; Gimenes-Fernandes, N.; Gottwald, T.R.; Bové, J.M. Spatial and temporal analyses of citrus sudden death as a tool to generate hypotheses concerning its etiology. Phytopathology 2003, 93, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Valencia, P.; Loeza-Kuk, E.; Mora-Aguilera, G.; Febres, V.; Ochoa-Martínez, D.; Gutiérrez-Espinosa, A.; Jesus-Junior, W.C.; Correia-Malvas, C.; Arno-Wulf, N. Population Structure of Citrus Tristeza Virus Isolates and Its Association. Agrociencia 2008, 42, 85–93. [Google Scholar]
- Targon, M.L.P.N.; Astúa-Monge, G.; Kishi, L.; Freitas-Astúa, J.; Souza, A.A.; Santos, F.A.; Muller, G.W.; Machado, M.A. Avaliação de haplótipos do CTV em plantas com sintomas de morte súbita dos citros por SSCP e seqüenciamento dos genes da p20 e p23. In Proceedings of the XXVI Congresso Paulista de Fitopatologia, Araras, Sao Paulo; 2003; p. 71. (In Portuguese). [Google Scholar]
- Maccheroni, W.; Alegria, M.C.; Greggio, C.C.; Piazza, J.P.; Kamla, R.F.; Zacharias, P.R.A.; Bar-Joseph, M.; Kitajima, E.W.; Assumpção, L.C.; Camarotte, G.; et al. Identification and genomic characterization of a new virus (Tymoviridae family) associated with citrus sudden death disease. J. Virol. 2005, 79, 3028–3037. [Google Scholar] [CrossRef] [PubMed]
- Nouri, S.; Salemb, N.; Nigga, J.C.; Falk, B.W. Diverse Array of New Viral Sequences Identified in Worldwide Populations of the Asian Citrus Psyllid (Diaphorina citri) Using Viral Metagenomics. J. Virol. 2016, 90, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.; Choi, H.; Cho, J.K.; Yoon, J.-Y.; Choi, S.-K.; Cho, W.K. In silico approach to reveal viral populations in grapevine cultivar Tannat using transcriptome data. Sci. Rep. 2015, 5, 15841. [Google Scholar] [CrossRef] [PubMed]
- Prabha, K.; Baranwal, V.K.; Jain, R.K. Applications of next generation high throughput sequencing technologies in characterization, discovery and molecular interaction of plant viruses. Ind. J. Virol. 2013, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Gao, S.; Hernandez, A.G.; Wechter, W.P.; Fei, Z.; Ling, K.-S. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS ONE 2012, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, B.; Freeborough, M.J.; Maree, H.J.; Celton, J.M.; Rees, D.J.; Burger, J.T. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Bekesiova, I.; Nap, J.P.; Mlynarova, L. Isolation of high quality DNA and RNA from leaves of the carnivorous plant Drosera rotundifolia. Plant Mol. Biol. Report. 2011, 17, 269–277. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- NCBI: National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 11 February 2016).
- SnapGene software. Available online: http://www.snapgene.com/ (accessed on 11 February 2016).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; Mcwilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Licciardello, G.; Scuderi, G.; Ferraro, R.; Giampetruzzi, A.; Russo, M.; Lombardo, A.; Raspagliesi, D.; Bar-Joseph, M.; Catara1, A. Deep sequencing and analysis of small RNAs in sweet orange grafted on sour orange infected with two Citrus tristeza virus isolates prevalent in Sicily. Arch. Virol. 2015, 160, 2583–2589. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y. Genomic Analysis for Different Strains of Citrus tristeza virus in Taiwan. Plant Pathol. Microbiol. submitted.
- Blitvich, B.J.; Firth, A.E. Insect-specific flaviviruses: A systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses 2015, 7, 1927–1959. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.-C.; Shia, M.; Tianc, J.-H.; Lind, X.-D.; Gaoa, D.-Y.; Hea, J.-R.; Wanga, J.-B.; Lia, C.-X.; Kanga, Y.-J.; Yuc, B.; et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [PubMed]
- TMHMM server. Available online: http://www.cbs.dtu.dk/services/TMHMM/ (accessed on 11 February 2016).
- Stach-Machado, D.R.; Peroni, L.A.; Dias, L.C.F.; Caporrino, M.C.; Müller, G.W.; Targon, M.L.P.N.; Machado, M.A. Characterization of monoclonal antibodies for identification of the severe strain of ‘Capão Bonito’ Citrus tristeza virus. In Proceedings of the XV Conference of the International Organization of Citrus Virologists; 2002; pp. 165–171. [Google Scholar]
- Mawassi, M.; Mietkiewska, E.; Gofman, R.; Yang, G.; Bar-Joseph, M. Unusual sequence relationships between two isolates of Citrus tristeza virus. J. Gen. Virol. 1996, 77, 2359–2364. [Google Scholar] [CrossRef] [PubMed]
- Suastika, G.; Natsuaki, T.; Terui, H.; Kano, T.; Ikei, H.; Okuda, S. Nucleotide sequence of Citrus tristeza virus seedling yellows isolate. J. Gen. Plant Pathol. 2001, 67, 73–77. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, S.; Moreno, P.; Guerri, J.; Ambros, S. The complete nucleotide sequence of a severe stem pitting isolate of Citrus tristeza virus from Spain: comparison with isolates from different origins. Arch. Virol. 2006, 151, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Dawson, T.E.; Pearson, M.N. Isolates of Citrus tristeza virus that overcome Poncirus trifoliata resistance comprise a novel strain. Arch. Virol. 2010, 155, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruiz, S.; Navarro, B.; Gisel, A.; Peña, L.; Navarro, L.; Moreno, P.; Di Serio, F.; Flores, R. Citrus tristeza virus infection induces the accumulation of viral small RNAs (21–24-nt) mapping preferentially at the 3′-terminal region of the genomic RNA and affects the host small RNA profile. Plant Mol. Biol. 2011, 75, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Albiach-marti, M.R. The Complex Genetics of Citrus tristeza virus. In Current Issues in Molecular Virology-Viral Genetics and Biotechnological Applications; Romanowski, V., Ed.; InTech: Rijeka, Croatia, 2013; pp. 1–26. [Google Scholar]
- Matsumura, E.E.; Coletta-Filho, H.D.; Dorta, S.O.; Nouri, S.; Machado, M.A. Genetic Structure and Molecular Variability Analysis of Citrus sudden death-associated virus Isolates from Infected Plants Grown in Brazil. Viruses 2016, 8, 330. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, B.E.; Menke, J.; Dahal, G.; Olszewski, N.E. Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Genet. Virol. 2000, 81, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Noreen, F.; Akbergenov, R.; Hohn, T.; Richert-Poggeler, K.R. Distinct expression of endogenous Petunia vein clearing virus and the DNA transposon dTph1 in two Petunia hybrida lines is correlated with differences in histone modification and siRNA production. Plant J. 2007, 50, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zavallo, D.; Debat, H.J.; Conti, G.; Manacorda, C.A.; Rodriguez, M.C.; Asurmendi, S. Differential mRNA accumulation upon early Arabidopsis thaliana infection with ORMV and TMV-Cg is associated with distinct endogenous small RNAs level. PLoS ONE 2015, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Canopy/Rootstock | Collected Tissue | Type of Library Constructed | Library ID | |
|---|---|---|---|---|
| Asymptomatic plants | 1 Valencia/Rough lemon | Roots | sRNA | SN453 |
| Valencia/Citrandarin Cleopatra × Rubidoux | Leaves | sRNA | SN468 | |
| 1 Valencia/Rough lemon | Leaves | sRNA | SN470 | |
| Valencia/Trifoliata Tristeno | Leaves | sRNA | SN473 | |
| Valencia/Rangpur lime × Swingle A | Leaves | sRNA | SN476 | |
| Valencia/Sunki mandarin | Leaves | sRNA | SN483 | |
| Valencia/Sunki × Cleopatra | Leaves | sRNA | SN486 | |
| Valencia/Swingle | Leaves | sRNA | SN488 | |
| 2 Valencia/Rangpur lime | Leaves | RNA-seq | C1-960 | |
| 2 Valencia/Rangpur lime | Roots | RNA-seq | C4-964 | |
| Valencia/Sunki of China | Leaves | RNA-seq | C1-963 | |
| Symptomatic plants | 3 Valencia/Rough lemon | Roots | sRNA | SN464 |
| 3 Valencia/Rough lemon | Leaves | sRNA | SN456 | |
| Valencia/Rangpur lime × Swingle A | Leaves | sRNA | SN459 | |
| Valencia/Citrus pennivesiculata | Leaves | sRNA | SN462 | |
| Valencia/Rangpur lime | Leaves | sRNA | SN479 | |
| 4 Valencia/Rangpur lime | Leaves | RNA-seq | C1-961 | |
| 4 Valencia/Rangpur lime | Roots | RNA-seq | C4-965 | |
| Valencia/Sunki of China | Leaves | RNA-seq | C1-962 | |
| Total of plants: 15; total of samples: 19 | ||||
| Library ID | No. of Reads after Trimming | No. of Exogenous Reads | 3 No. of Putative Viral Contigs Detected in an Overall Screening | 4 No. of Viral Contigs Detected in an Individual Screening | ||
|---|---|---|---|---|---|---|
| ≤200 nt | Between 201 to 999 nt | ≥1000 nt | ||||
| 1 C1-960 | 37,811,400 | 3,826,317 | 40,187 | 76 | 41 | 7 |
| 1 C1-961 | 37,380,448 | 3,613,441 | 29,483 | 74 | 37 | 9 |
| 1 C1-962 | 36,452,005 | 3,528,784 | 30,834 | 72 | 32 | 6 |
| 1 C1-963 | 29,942,484 | 3,027,793 | 38,028 | 54 | 25 | 2 |
| 1 C4-964 | 35,511,705 | 3,741,954 | 33,719 | 10 | 4 | 4 |
| 1 C4-965 | 35,386,080 | 3,617,222 | 39,012 | 24 | 13 | 0 |
| 2 SN453 | 8,091,654 | 756,302 | 776 | 323 | 19 | 0 |
| 2 SN456 | 8,949,837 | 1,117,451 | 395 | 263 | 5 | 0 |
| 2 SN459 | 9,899,316 | 1,065,849 | 571 | 334 | 6 | 0 |
| 2 SN462 | 6,233,982 | 836,217 | 267 | 198 | 5 | 0 |
| 2 SN464 | 9,042,291 | 782,698 | 821 | 178 | 29 | 0 |
| 2 SN468 | 8,843,660 | 897,286 | 626 | 384 | 11 | 0 |
| 2 SN470 | 6,866,756 | 1,041,001 | 410 | 329 | 4 | 0 |
| 2 SN473 | 11,892,105 | 1,597,924 | 533 | 311 | 41 | 0 |
| 2 SN476 | 9,076,165 | 917,605 | 631 | 408 | 14 | 0 |
| 2 SN479 | 11,649,840 | 1,232,264 | 733 | 163 | 24 | 0 |
| 2 SN483 | 8,765,391 | 949,445 | 471 | 326 | 13 | 0 |
| 2 SN486 | 11,001,152 | 1,127,439 | 555 | 242 | 13 | 0 |
| 2 SN488 | 14,231,970 | 1,446,481 | 783 | 507 | 20 | 0 |
| Total | 337,028,241 | 35,123,473 | 218,835 | 4276 | 356 | 28 |
| Closely Related Viruses | Family | No. of Contigs | Contigs Length (nt) | From RNA-Seq Libraries | From sRNA Libraries | Maximum % aa Identity |
|---|---|---|---|---|---|---|
| Citrus tristeza virus | Closteroviridae | 4556 | 50–3180 | 560 | 3996 | 92 |
| Citrus sudden death-associated virus | Tymoviridae | 61 | 50–6109 | 20 | 41 | 98 |
| Marine RNA virus SF-2 | Marnaviridae | 1 | 1400 | 1 | 0 | 22 |
| Po-Circo-like virus 51 | Circoviridae | 1 | 305 | 1 | 0 | 43 |
| Aphid lethal paralysis virus | Dicistroviridae | 6 | 115–343 | 6 | 0 | 97 |
| Nakiwogo virus | Flaviviridae | 1 | 2512 | 1 | 0 | 27 |
| Sclerotinia sclerotiorum deltaflexivirus 1 | Flexiviridae | 5 | 101–329 | 5 | 0 | 62 |
| Citrus endogenous pararetrovirus | Caulimoviridae | 8 | 339–3339 | 8 | 0 | 72 |
| Fusarium graminearum deltaflexivirus 1 | Putative Deltaflexiviridae | 2 | 153–262 | 2 | 0 | 71 |
| Boutonnet virus | unclassified viruses | 2 | 423–434 | 2 | 0 | 36 |
| Beet virus Q | Virgaviridae | 1 | 4097 | 1 | 0 | 33 |
| Chinese wheat mosaic virus | Virgaviridae | 1 | 2626 | 1 | 0 | 28 |
| Virus | Reference Isolate | sRNA Simultaneous Re-Assembly | RNA-Seq Simultaneous Re-Assembly | ||||
|---|---|---|---|---|---|---|---|
| Read Count | Percentage Read Count | Average Coverage | Read Count | Percentage Read Count | Average Coverage | ||
| CTV | A18 | 711,217 | 15.8% | ≈740× | 2,450 | 12.8% | ≈13× |
| Taiwan-Pum | 1,800,699 | 40.1% | ≈1870× | 6,789 | 35.5% | ≈35× | |
| SG29 | 1,980,214 | 44.1% | ≈2060× | 9,882 | 51.7% | ≈50× | |
| Total | 4,492,130 | 100% | − | 19,121 | 100% | − | |
| CSDaV | AY884005 | 3944 | 69.6% | ≈12× | 59,916 | 73.3% | ≈810× |
| DQ185573 | 1723 | 30.4% | ≈5× | 21,784 | 26.7% | ≈295× | |
| Total | 5667 | 100% | − | 81,700 | 100% | − | |
| 1 Reference Viral/Contig Sequence | sRNA Simultaneous Re-Assembly | RNA-Seq Simultaneous Re-Assembly | ||
|---|---|---|---|---|
| Read Count | Average Coverage | Read Count | Average Coverage | |
| ALPV | 387 | 0.52x | 113 | 1.1x |
| CitPRV | 21,693 | 68.22x | 2196 | 28.88x |
| SsDFV1 | 227 | 0.49x | 3 | 0.02x |
| CtgCirco-1 | 12 | 0.53x | 25 | 6.57x |
| CtgFlavi-1 | 89 | 0.53x | 3144 | 113.95x |
| CtgMarna-1 | 68 | 0.64x | 83 | 5.82x |
| CtgUnclass-1 | 103 | 3.37x | 189 | 41.4x |
| CtgVirga-1 | 163 | 0.59x | 2297 | 51.4x |
| CtgVirga-2 | 105 | 0.57x | 1723 | 61.8x |
| Reference Viral Sequence | Asymptomatic Libraries Re-Assembly | Symptomatic Libraries Re-Assembly | ||
|---|---|---|---|---|
| Read Count | Average Coverage | Read Count | Average Coverage | |
| CTV_SPBR_01 | 418,902 | 442.32x | 525,380 | 563.19x |
| CTV_SPBR_02 | 553,150 | 584.91x | 633,371 | 693.78x |
| CSDaV_SPBR_01 | 3934 | 26.49x | 58,532 | 767.43x |
| CSDaV_SPBR_02 | 8844 | 109.53x | 14,350 | 182.44x |
| CtgFlavi-1 | 3182 | 114.2x | 28 | 0.15x |
| CtgVirga-1 | 2582 | 56.14x | 59 | 0.2x |
| CtgVirga-2 | 1791 | 62.18x | 12 | 0.06x |
| CitPRV_SPBR_01 | 721 | 8.35x | 8325 | 41.65x |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumura, E.E.; Coletta-Filho, H.D.; Nouri, S.; Falk, B.W.; Nerva, L.; Oliveira, T.S.; Dorta, S.O.; Machado, M.A. Deep Sequencing Analysis of RNAs from Citrus Plants Grown in a Citrus Sudden Death-Affected Area Reveals Diverse Known and Putative Novel Viruses. Viruses 2017, 9, 92. https://doi.org/10.3390/v9040092
Matsumura EE, Coletta-Filho HD, Nouri S, Falk BW, Nerva L, Oliveira TS, Dorta SO, Machado MA. Deep Sequencing Analysis of RNAs from Citrus Plants Grown in a Citrus Sudden Death-Affected Area Reveals Diverse Known and Putative Novel Viruses. Viruses. 2017; 9(4):92. https://doi.org/10.3390/v9040092
Chicago/Turabian StyleMatsumura, Emilyn E., Helvecio D. Coletta-Filho, Shahideh Nouri, Bryce W. Falk, Luca Nerva, Tiago S. Oliveira, Silvia O. Dorta, and Marcos A. Machado. 2017. "Deep Sequencing Analysis of RNAs from Citrus Plants Grown in a Citrus Sudden Death-Affected Area Reveals Diverse Known and Putative Novel Viruses" Viruses 9, no. 4: 92. https://doi.org/10.3390/v9040092
APA StyleMatsumura, E. E., Coletta-Filho, H. D., Nouri, S., Falk, B. W., Nerva, L., Oliveira, T. S., Dorta, S. O., & Machado, M. A. (2017). Deep Sequencing Analysis of RNAs from Citrus Plants Grown in a Citrus Sudden Death-Affected Area Reveals Diverse Known and Putative Novel Viruses. Viruses, 9(4), 92. https://doi.org/10.3390/v9040092