
Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift?


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Human Papillomavirus Replication
3. Human Papillomavirus Replication and the DNA Damage Response: Primed for Integration
4. Human Papillomavirus and Integration in Cervical Cancer
5. Human Papillomavirus and Head and Neck Cancer
6. Human Papillomavirus and Integration in Head and Neck Cancer
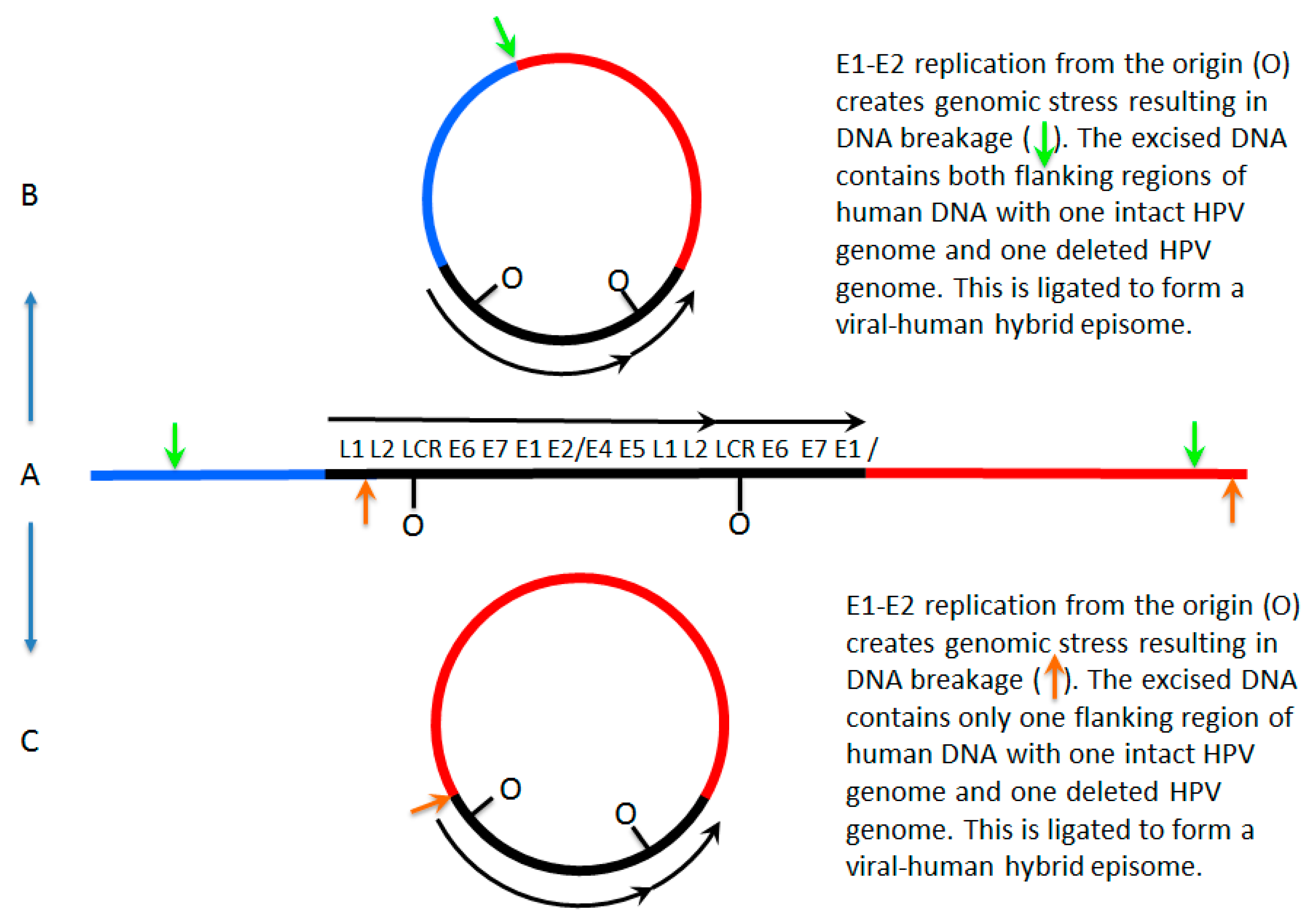
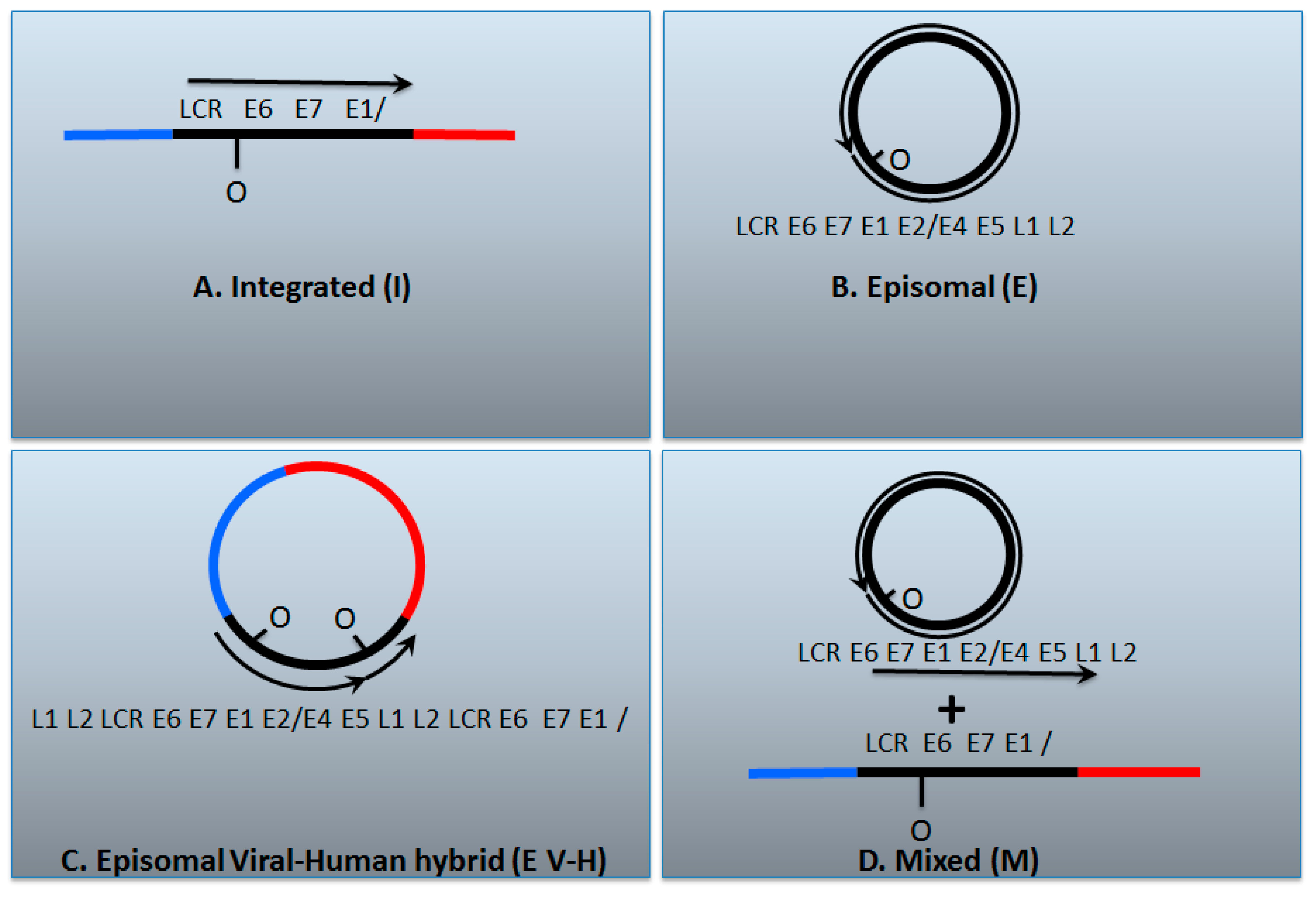
7. A Model for Integration and Excision of HPV DNA
8. Conflicting Interpretations Rather Than Conflicting Results
9. Why This Matters: De-Escalation Therapy for Human Papillomavirus-Positive Head and Neck Cancer
10. Future Approaches to the Diagnostic Management of Human Papillomavirus-Positive Head and Neck Cancer
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nulton, T.J.; Olex, A.L.; Dozmorov, M.; Morgan, I.M.; Windle, B. Analysis of the Cancer Genome Atlas Sequencing Data Reveals Novel Properties of the Human Papillomavirus 16 Genome in Head and Neck Squamous Cell Carcinoma. Oncotarget 2017, 8, 17684–17699. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.M.; Laimins, L.A. Human Papillomaviruses: Basic Mechanisms of Pathogenesis and Oncogenicity. Rev. Med. Virol. 2006, 16, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the Papillomavirus L2/vDNA Complex Across the Limiting Membrane Requires the Onset of Mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kuhbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large Scale RNAi Reveals the Requirement of Nuclear Envelope Breakdown for Nuclear Import of Human Papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F. Transcriptional Regulation of the Papillomavirus Oncogenes by Cellular and Viral Transcription Factors in Cervical Carcinoma. Virology 2009, 384, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomavirus Oncoproteins: Pathways to Transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The Papillomavirus E2 Proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 Proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Masterson, P.J.; Stanley, M.A.; Lewis, A.P.; Romanos, M.A. A C-Terminal Helicase Domain of the Human Papillomavirus E1 Protein Binds E2 and the DNA Polymerase Alpha-Primase p68 Subunit. J. Virol. 1998, 72, 7407–7419. [Google Scholar] [PubMed]
- Melendy, T.; Sedman, J.; Stenlund, A. Cellular Factors Required for Papillomavirus DNA Replication. J. Virol. 1995, 69, 7857–7867. [Google Scholar] [PubMed]
- Hu, Y.; Clower, R.V.; Melendy, T. Cellular Topoisomerase I Modulates Origin Binding by Bovine Papillomavirus Type 1 E1. J. Virol. 2006, 80, 4363–4371. [Google Scholar] [CrossRef] [PubMed]
- Clower, R.V.; Fisk, J.C.; Melendy, T. Papillomavirus E1 Protein Binds to and Stimulates Human Topoisomerase I. J. Virol. 2006, 80, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Melendy, T. Recruitment of Replication Protein A by the Papillomavirus E1 Protein and Modulation by Single-Stranded DNA. J. Virol. 2004, 78, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A Functional Interaction between the Human Papillomavirus 16 Transcription/Replication Factor E2 and the DNA Damage Response Protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Mackintosh, L.J.; Bodily, J.M.; Dornan, E.S.; Laimins, L.A.; Morgan, I.M. An Interaction between Human Papillomavirus 16 E2 and TopBP1 is Required for Optimum Viral DNA Replication and Episomal Genome Establishment. J. Virol. 2012, 86, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence Supporting a Role for TopBP1 and Brd4 in the Initiation but Not Continuation of Human Papillomavirus 16 E1/E2-Mediated DNA Replication. J. Virol. 2015, 89, 17684–17699. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Boner, W.; Morgan, I.M. TopBP1 Regulates Human Papillomavirus Type 16 E2 Interaction with Chromatin. J. Virol. 2007, 81, 4338–4342. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the Bovine Papillomavirus E2 Protein with Brd4 Tethers the Viral DNA to Host Mitotic Chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Parish, J.L.; Bean, A.M.; Park, R.B.; Androphy, E.J. ChlR1 is Required for Loading Papillomavirus E2 Onto Mitotic Chromosomes and Viral Genome Maintenance. Mol. Cell 2006, 24, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Gautam, D.; Ok, S.T.; Johnson, B.A.; Anacker, D.C.; Moody, C.A. Homologous Recombination Repair Factors, Rad51 and BRCA1, are Necessary for Productive Replication of Human Papillomavirus 31. J. Virol. 2015, 90, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human Papillomaviruses Recruit Cellular DNA Repair and Homologous Recombination Factors to Viral Replication Centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Moody, C.A.; Weitzman, M.D.; Weitzman, J.B.; Moody, C.A.; Laimins, L.A.; Hong, S.; Dutta, A.; Laimins, L.A.; Gillespie, K.A.; et al. Impact of the DNA Damage Response on Human Papillomavirus Chromatin. PLoS Pathog. 2016, 12, e1005613. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear Accumulation of the Papillomavirus E1 Helicase Blocks S-Phase Progression and Triggers an ATM-Dependent DNA Damage Response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive Replication of Human Papillomavirus 31 Requires DNA Repair Factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Kim, B.; Moody, C.A. HPV31 Utilizes the ATR-Chk1 Pathway to Maintain Elevated RRM2 Levels and a Replication-Competent Environment in Differentiating Keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Aloor, H.L.; Moody, C.A. The Rb Binding Domain of HPV31 E7 is Required to Maintain High Levels of DNA Repair Factors in Infected Cells. Virology 2017, 500, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Bristol, M.L.; Wang, X.; Smith, N.W.; Son, M.P.; Evans, M.R.; Morgan, I.M. DNA Damage Reduces the Quality, but Not the Quantity of Human Papillomavirus 16 E1 and E2 DNA Replication. Viruses 2016, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-Dependent DNA Damage Response at the HPV18 Replication Centers during the Initial Amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The Papillomavirus E1 Helicase Activates a Cellular DNA Damage Response in Viral Replication Foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.Y.; Chiang, C.M.; McBride, A.A. Brd4 is Displaced from HPV Replication Factories as they Expand and Amplify Viral DNA. PLoS Pathog. 2013, 9, e1003777. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use Recombination-Dependent Replication to Vegetatively Amplify their Genomes in Differentiated Cells. PLoS Pathog. 2013, 9, e1003321. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Fisk, J.C.; Dornan, E.S.; Donaldson, M.M.; Melendy, T.; Morgan, I.M. Human Papillomavirus E1 and E2 Mediated DNA Replication is Not Arrested by DNA Damage Signalling. Virology 2010, 406, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the Causation of Human Cancers—A Brief Historical Account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Cullen, A.P.; Reid, R.; Campion, M.; Lorincz, A.T. Analysis of the Physical State of Different Human Papillomavirus DNAs in Intraepithelial and Invasive Cervical Neoplasm. J. Virol. 1991, 65, 606–612. [Google Scholar] [PubMed]
- Durst, M.; Kleinheinz, A.; Hotz, M.; Gissmann, L. The Physical State of Human Papillomavirus Type 16 DNA in Benign and Malignant Genital Tumours. J. Gen. Virol. 1985, 66, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, E.; Jenkins, A.; Holm, R. Coexistence of Episomal and Integrated HPV16 DNA in Squamous Cell Carcinoma of the Cervix. J. Clin. Pathol. 1994, 47, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of Human Papillomavirus Type 16 into the Human Genome Correlates with a Selective Growth Advantage of Cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Cooper, K.; Herrington, C.S.; Lo, E.S.; Evans, M.F.; McGee, J.O. Integration of Human Papillomavirus Types 16 and 18 in Cervical Adenocarcinoma. J. Clin. Pathol. 1992, 45, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Sastre-Garau, X.; Schneider-Maunoury, S.; Labib, A.; Orth, G. Integration of Papillomavirus DNA near Myc Genes in Genital Carcinomas and its Consequences for Proto-Oncogene Expression. J. Virol. 1991, 65, 4534–4538. [Google Scholar] [PubMed]
- Cooper, K.; Herrington, C.S.; Graham, A.K.; Evans, M.F.; McGee, J.O. In Situ Human Papillomavirus (HPV) Genotyping of Cervical Intraepithelial Neoplasia in South African and British Patients: Evidence for Putative HPV Integration in Vivo. J. Clin. Pathol. 1991, 44, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, M.; Yamakawa, Y.; Shimano, S.; Hashimoto, M.; Sawada, Y.; Fujinaga, K. The Physical State of Human Papillomavirus 16 DNA in Cervical Carcinoma and Cervical Intraepithelial Neoplasia. Cancer 1990, 66, 2155–2161. [Google Scholar] [CrossRef]
- Wagatsuma, M.; Hashimoto, K.; Matsukura, T. Analysis of Integrated Human Papillomavirus Type 16 DNA in Cervical Cancers: Amplification of Viral Sequences Together with Cellular Flanking Sequences. J. Virol. 1990, 64, 813–821. [Google Scholar] [PubMed]
- Woodworth, C.D.; Bowden, P.E.; Doniger, J.; Pirisi, L.; Barnes, W.; Lancaster, W.D.; DiPaolo, J.A. Characterization of Normal Human Exocervical Epithelial Cells Immortalized in Vitro by Papillomavirus Types 16 and 18 DNA. Cancer Res. 1988, 48, 4620–4628. [Google Scholar] [PubMed]
- Lehn, H.; Villa, L.L.; Marziona, F.; Hilgarth, M.; Hillemans, H.G.; Sauer, G. Physical State and Biological Activity of Human Papillomavirus Genomes in Precancerous Lesions of the Female Genital Tract. J. Gen. Virol. 1988, 69, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.B.; Pan, C.C.; Han, S.H. Integration of Human Papillomavirus Type 16 into Cellular DNA of Cervical Carcinoma: Preferential Deletion of the E2 Gene and Invariable Retention of the Long Control Region and the E6/E7 Open Reading Frames. Virology 1987, 161, 259–261. [Google Scholar] [CrossRef]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and Transcriptional Analysis of Human Papillomavirus Type 16 Sequences in Cervical Carcinoma Cell Lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [PubMed]
- Durst, M.; Croce, C.M.; Gissmann, L.; Schwarz, E.; Huebner, K. Papillomavirus Sequences Integrate Near Cellular Oncogenes in some Cervical Carcinomas. Proc. Natl. Acad. Sci. USA 1987, 84, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Choo, K.B.; Pan, C.C.; Liu, M.S.; Ng, H.T.; Chen, C.P.; Lee, Y.N.; Chao, C.F.; Meng, C.L.; Yeh, M.Y.; Han, S.H. Presence of Episomal and Integrated Human Papillomavirus DNA Sequences in Cervical Carcinoma. J. Med. Virol. 1987, 21, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, H.; Tomita, Y.; Sekiya, S.; Takamizawa, H.; Simizu, B. Integration and Transcription of Human Papillomavirus Type 16 and 18 Sequences in Cell Lines Derived from Cervical Carcinomas. J. Gen. Virol. 1987, 68, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Mincheva, A.; Gissmann, L.; zur Hausen, H. Chromosomal Integration Sites of Human Papillomavirus DNA in Three Cervical Cancer Cell Lines Mapped by in Situ Hybridization. Med. Microbiol. Immunol. 1987, 176, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Romanczuk, H.; Howley, P.M. Disruption of either the E1 or the E2 Regulatory Gene of Human Papillomavirus Type 16 Increases Viral Immortalization Capacity. Proc. Natl. Acad. Sci. USA 1992, 89, 3159–3163. [Google Scholar] [CrossRef] [PubMed]
- Daniel, B.; Mukherjee, G.; Seshadri, L.; Vallikad, E.; Krishna, S. Changes in the Physical State and Expression of Human Papillomavirus Type 16 in the Progression of Cervical Intraepithelial Neoplasia Lesions Analysed by PCR. J. Gen. Virol. 1995, 76 Pt 10, 2589–2593. [Google Scholar] [CrossRef] [PubMed]
- Vernon, S.D.; Unger, E.R.; Miller, D.L.; Lee, D.R.; Reeves, W.C. Association of Human Papillomavirus Type 16 Integration in the E2 Gene with Poor Disease-Free Survival from Cervical Cancer. Int. J. Cancer 1997, 74, 50–56. [Google Scholar] [CrossRef]
- Park, J.S.; Hwang, E.S.; Park, S.N.; Ahn, H.K.; Um, S.J.; Kim, C.J.; Kim, S.J.; Namkoong, S.E. Physical Status and Expression of HPV Genes in Cervical Cancers. Gynecol. Oncol. 1997, 65, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Yoshinouchi, M.; Hongo, A.; Nakamura, K.; Kodama, J.; Itoh, S.; Sakai, H.; Kudo, T. Analysis by Multiplex PCR of the Physical Status of Human Papillomavirus Type 16 DNA in Cervical Cancers. J. Clin. Microbiol. 1999, 37, 3514–3517. [Google Scholar] [PubMed]
- Gray, E.; Pett, M.R.; Ward, D.; Winder, D.M.; Stanley, M.A.; Roberts, I.; Scarpini, C.G.; Coleman, N. In Vitro Progression of Human Papillomavirus 16 Episome-Associated Cervical Neoplasia Displays Fundamental Similarities to Integrant-Associated Carcinogenesis. Cancer Res. 2010, 70, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Herdman, M.T.; Pett, M.R.; Roberts, I.; Alazawi, W.O.F.; Teschendorff, A.E.; Zhang, X.; Stanley, M.A.; Coleman, N. Interferon-Beta Treatment of Cervical Keratinocytes Naturally Infected with Human Papillomavirus 16 Episomes Promotes Rapid Reduction in Episome Numbers and Emergence of Latent Integrants. Carcinogenesis 2006, 27, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Storey, A.; Pim, D.; Banks, L. Characterization of the Human Papillomavirus E2 Protein: Evidence of Trans-Activation and Trans-Repression in Cervical Keratinocytes. EMBO J. 1994, 13, 5451–5459. [Google Scholar] [PubMed]
- Demeret, C.; Desaintes, C.; Yaniv, M.; Thierry, F. Different Mechanisms Contribute to the E2-Mediated Transcriptional Repression of Human Papillomavirus Type 18 Viral Oncogenes. J. Virol. 1997, 71, 9343–9349. [Google Scholar] [PubMed]
- Romanczuk, H.; Thierry, F.; Howley, P.M. Mutational Analysis of cis Elements Involved in E2 Modulation of Human Papillomavirus Type 16 P97 and Type 18 P105 Promoters. J. Virol. 1990, 64, 2849–2859. [Google Scholar] [PubMed]
- Bechtold, V.; Beard, P.; Raj, K. Human Papillomavirus Type 16 E2 Protein has no Effect on Transcription from Episomal Viral DNA. J. Virol. 2003, 77, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Naeger, L.K.; DiMaio, D. Activation of the Endogenous p53 Growth Inhibitory Pathway in HeLa Cervical Carcinoma Cells by Expression of the Bovine Papillomavirus E2 Gene. Oncogene 1996, 12, 795–803. [Google Scholar] [PubMed]
- DeFilippis, R.A.; Goodwin, E.C.; Wu, L.; DiMaio, D. Endogenous Human Papillomavirus E6 and E7 Proteins Differentially Regulate Proliferation, Senescence, and Apoptosis in HeLa Cervical Carcinoma Cells. J. Virol. 2003, 77, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; DiMaio, D. Induced Senescence in HeLa Cervical Carcinoma Cells Containing Elevated Telomerase Activity and Extended Telomeres. Cell Growth Differ. 2001, 12, 525–534. [Google Scholar] [PubMed]
- Parish, J.L.; Kowalczyk, A.; Chen, H.; Roeder, G.E.; Sessions, R.; Buckle, M.; Gaston, K. E2 Proteins from High- and Low-Risk Human Papillomavirus Types Differ in their Ability to Bind p53 and Induce Apoptotic Cell Death E2 Proteins from High- and Low-Risk Human Papillomavirus Types Differ in their Ability to Bind p53 and Induce Apoptotic Cell. J. Virol. 2006, 80, 4580–4590. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, M.; Bernard, H.U. Assessment of the HPV DNA Methylation Status in Cervical Lesions. Methods Mol. Biol. 2015, 1249, 267–280. [Google Scholar]
- Kalantari, M.; Calleja-Macias, I.E.; Tewari, D.; Hagmar, B.; Lie, K.; Barrera-Saldana, H.A.; Wiley, D.J.; Bernard, H.U. Conserved Methylation Patterns of Human Papillomavirus Type 16 DNA in Asymptomatic Infection and Cervical Neoplasia. J. Virol. 2004, 78, 12762–12772. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Bhattacharjee, B.; Sen, S.; Mukhopadhyay, I.; Sengupta, S. Association of Viral Load with HPV16 Positive Cervical Cancer Pathogenesis: Causal Relevance in Isolates Harboring Intact Viral E2 Gene. Virology 2010, 402, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, B.; Sengupta, S. CpG Methylation of HPV 16 LCR at E2 Binding Site Proximal to P97 is Associated with Cervical Cancer in Presence of Intact E2. Virology 2006, 354, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Rosales, C.; Lopez-Nieva, P.; Grana, O.; Ballestar, E.; Ropero, S.; Espada, J.; Melo, S.A.; Lujambio, A.; Fraga, M.F.; et al. The Dynamic DNA Methylomes of Double-Stranded DNA Viruses Associated with Human Cancer. Genome Res. 2009, 19, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Sun, C.; Ghosh, A.; Kinney, W.; Mirabello, L.; Wacholder, S.; Shaber, R.; LaMere, B.; Clarke, M.; Lorincz, A.T.; et al. Methylation of HPV18, HPV31, and HPV45 Genomes and Cervical Intraepithelial Neoplasia Grade 3. J. Natl. Cancer Inst. 2012, 104, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Badal, V.; Chuang, L.S.; Tan, E.H.; Badal, S.; Villa, L.L.; Wheeler, C.M.; Li, B.F.; Bernard, H.U. CpG Methylation of Human Papillomavirus Type 16 DNA in Cervical Cancer Cell Lines and in Clinical Specimens: Genomic Hypomethylation Correlates with Carcinogenic Progression. J. Virol. 2003, 77, 6227–6234. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, M.; Lee, D.; Calleja-Macias, I.E.; Lambert, P.F.; Bernard, H.U. Effects of Cellular Differentiation, Chromosomal Integration and 5-aza-2′-Deoxycytidine Treatment on Human Papillomavirus-16 DNA Methylation in Cultured Cell Lines. Virology 2008, 374, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a Causal Association between Human Papillomavirus and a Subset of Head and Neck Cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L. Human Papillomavirus-Associated Head and Neck Cancer is a Distinct Epidemiologic, Clinical, and Molecular Entity. Semin. Oncol. 2004, 31, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Shah, K.V. Human Papillomavirus-Associated Head and Neck Squamous Cell Carcinoma: Mounting Evidence for an Etiologic Role for Human Papillomavirus in a Subset of Head and Neck Cancers. Curr. Opin. Oncol. 2001, 13, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Garnaes, E.; Kiss, K.; Andersen, L.; Therkildsen, M.H.; Franzmann, M.B.; Filtenborg-Barnkob, B.; Hoegdall, E.; Lajer, C.B.; Andersen, E.; Specht, L.; et al. Increasing Incidence of Base of Tongue Cancers from 2000 to 2010 due to HPV: The Largest Demographic Study of 210 Danish Patients. Br. J. Cancer 2015, 113, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Garnaes, E.; Kiss, K.; Andersen, L.; Therkildsen, M.H.; Franzmann, M.B.; Filtenborg-Barnkob, B.; Hoegdall, E.; Krenk, L.; Josiassen, M.; Lajer, C.B.; et al. A High and Increasing HPV Prevalence in Tonsillar Cancers in Eastern Denmark, 2000–2010: The Largest Registry-Based Study to Date. Int. J. Cancer 2015, 136, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Carlander, A.F.; Gronhoj Larsen, C.; Jensen, D.H.; Garnaes, E.; Kiss, K.; Andersen, L.; Olsen, C.H.; Franzmann, M.; Hogdall, E.; Kjaer, S.K.; et al. Continuing Rise in Oropharyngeal Cancer in a High HPV Prevalence Area: A Danish Population-Based Study from 2011 to 2014. Eur. J. Cancer 2017, 70, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human Papillomavirus and Survival of Patients with Oropharyngeal Cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Garnaes, E.; Frederiksen, K.; Kiss, K.; Andersen, L.; Therkildsen, M.H.; Franzmann, M.B.; Specht, L.; Andersen, E.; Norrild, B.; Kjaer, S.K.; et al. Double Positivity for HPV DNA/p16 in Tonsillar and Base of Tongue Cancer Improves Prognostication: Insights from a Large Population-Based Study. Int. J. Cancer 2016, 139, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.G.; Jensen, D.H.; Carlander, A.F.; Kiss, K.; Andersen, L.; Olsen, C.H.; Andersen, E.; Garnaes, E.; Cilius, F.; Specht, L.; et al. Novel Nomograms for Survival and Progression in HPV+ and HPV− Oropharyngeal Cancer: A Population-Based Study of 1542 Consecutive Patients. Oncotarget 2016, 7, 71761–71772. [Google Scholar] [PubMed]
- Hafkamp, H.C.; Speel, E.J.; Haesevoets, A.; Bot, F.J.; Dinjens, W.N.; Ramaekers, F.C.; Hopman, A.H.; Manni, J.J. A Subset of Head and Neck Squamous Cell Carcinomas Exhibits Integration of HPV 16/18 DNA and Overexpression of p16INK4A and p53 in the Absence of Mutations in p53 Exons 5–8. Int. J. Cancer 2003, 107, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Begum, S.; Cao, D.; Gillison, M.; Zahurak, M.; Westra, W.H. Tissue Distribution of Human Papillomavirus 16 DNA Integration in Patients with Tonsillar Carcinoma. Clin. Cancer Res. 2005, 11, 5694–5699. [Google Scholar] [CrossRef] [PubMed]
- Samama, B.; Plas-Roser, S.; Schaeffer, C.; Chateau, D.; Fabre, M.; Boehm, N. HPV DNA Detection by in Situ Hybridization with Catalyzed Signal Amplification on Thin-Layer Cervical Smears. J. Histochem. Cytochem. 2002, 50, 1417–1420. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Koo, B.S.; Kang, S.; Park, K.; Kim, H.; Lee, K.R.; Lee, M.J.; Kim, J.M.; Choi, E.C.; Cho, N.H. HPV Integration Begins in the Tonsillar Crypt and Leads to the Alteration of p16, EGFR and c-Myc during Tumor Formation. Int. J. Cancer 2007, 120, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Hasegawa, M.; Kiyuna, A.; Matayoshi, S.; Uehara, T.; Agena, S.; Yamashita, Y.; Ogawa, K.; Maeda, H.; Suzuki, M. Viral Load, Physical Status, and E6/E7 mRNA Expression of Human Papillomavirus in Head and Neck Squamous Cell Carcinoma. Head Neck 2013, 35, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Olthof, N.C.; Speel, E.M.; Kolligs, J.; Haesevoets, A.; Henfling, M.; Ramaekers, F.C.S.; Preuss, S.F.; Drebber, U.; Wieland, U.; Silling, S.; et al. Comprehensive Analysis of HPV16 Integration in OSCC Reveals no Significant Impact of Physical Status on Viral Oncogene and Virally Disrupted Human Gene Expression. PLoS ONE 2014, 9, e88718. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and Host Genome Interactions in Primary Head and Neck Cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Vojtechova, Z.; Sabol, I.; Salakova, M.; Turek, L.; Grega, M.; Smahelova, J.; Vencalek, O.; Lukesova, E.; Klozar, J.; Tachezy, R. Analysis of the Integration of Human Papillomaviruses in Head and Neck Tumours in Relation to Patients’ Prognosis. Int. J. Cancer 2016, 138, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Dahlstrom, K.R.; Sturgis, E.M.; Li, G. Human Papillomavirus Integration Pattern and Demographic, Clinical, and Survival Characteristics of Patients with Oropharyngeal Squamous Cell Carcinoma. Head Neck 2016, 38, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic Instability of the Host Cell Induced by the Human Papillomavirus Replication Machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-Wide Analysis of HPV Integration in Human Cancers Reveals Recurrent, Focal Genomic Instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.R.; James, C.D.; Loughran, O.; Nulton, T.J.; Wang, X.; Bristol, M.L.; Windle, B.; Morgan, I.M. An Oral Keratinocyte Life Cycle Model Identifies Novel Host Genome Regulation by Human Papillomavirus 16 Relevant to HPV Positive Head and Neck Cancer. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R.; Husain, Z.A.; Burtness, B. Treatment De-Intensification Strategies for Head and Neck Cancer. Eur. J. Cancer 2016, 68, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, C.; Westra, W.H.; Li, S.; Cmelak, A.; Ridge, J.A.; Pinto, H.; Forastiere, A.; Gillison, M.L. Improved Survival of Patients with Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma in a Prospective Clinical Trial. J. Natl. Cancer Inst. 2008, 100, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.A.; Kampmann, M.; Zlobec, I.; Green, E.; Tornillo, L.; Lugli, A.; Wolfensberger, M.; Terracciano, L.M. P16 Expression in Oropharyngeal Cancer: Its Impact on Staging and Prognosis Compared with the Conventional Clinical Staging Parameters. Ann. Oncol. 2010, 21, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Haughey, B.H.; Sinha, P.; Kallogjeri, D.; Goldberg, R.L.; Lewis, J.S., Jr.; Piccirillo, J.F.; Jackson, R.S.; Moore, E.J.; Brandwein-Gensler, M.; Magnuson, S.J.; et al. Pathology-Based Staging for HPV-Positive Squamous Carcinoma of the Oropharynx. Oral Oncol. 2016, 62, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lydiatt, W.M.; Patel, S.G.; O’Sullivan, B.; Brandwein, M.S.; Ridge, J.A.; Migliacci, J.C.; Loomis, A.M.; Shah, J.P. Head and Neck Cancers-Major Changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer. J. Clin. 2017, 67, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Malm, I.J.; Fan, C.J.; Yin, L.X.; Li, D.X.; Koch, W.M.; Gourin, C.G.; Pitman, K.T.; Richmon, J.D.; Westra, W.H.; Kang, H.; et al. Evaluation of Proposed Staging Systems for Human Papillomavirus-Related Oropharyngeal Squamous Cell Carcinoma. Cancer 2017, 123, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Porceddu, S.V.; Milne, R.; Brown, E.; Bernard, A.; Rahbari, R.; Cartmill, B.; Foote, M.; McGrath, M.; Coward, J.; Panizza, B. Validation of the ICON-S Staging for HPV-Associated Oropharyngeal Carcinoma using a Pre-Defined Treatment Policy. Oral Oncol. 2017, 66, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Ramqvist, T.; Mints, M.; Tertipis, N.; Nasman, A.; Romanitan, M.; Dalianis, T. Studies on Human Papillomavirus (HPV) 16 E2, E5 and E7 mRNA in HPV-Positive Tonsillar and Base of Tongue Cancer in Relation to Clinical Outcome and Immunological Parameters. Oral Oncol. 2015, 51, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
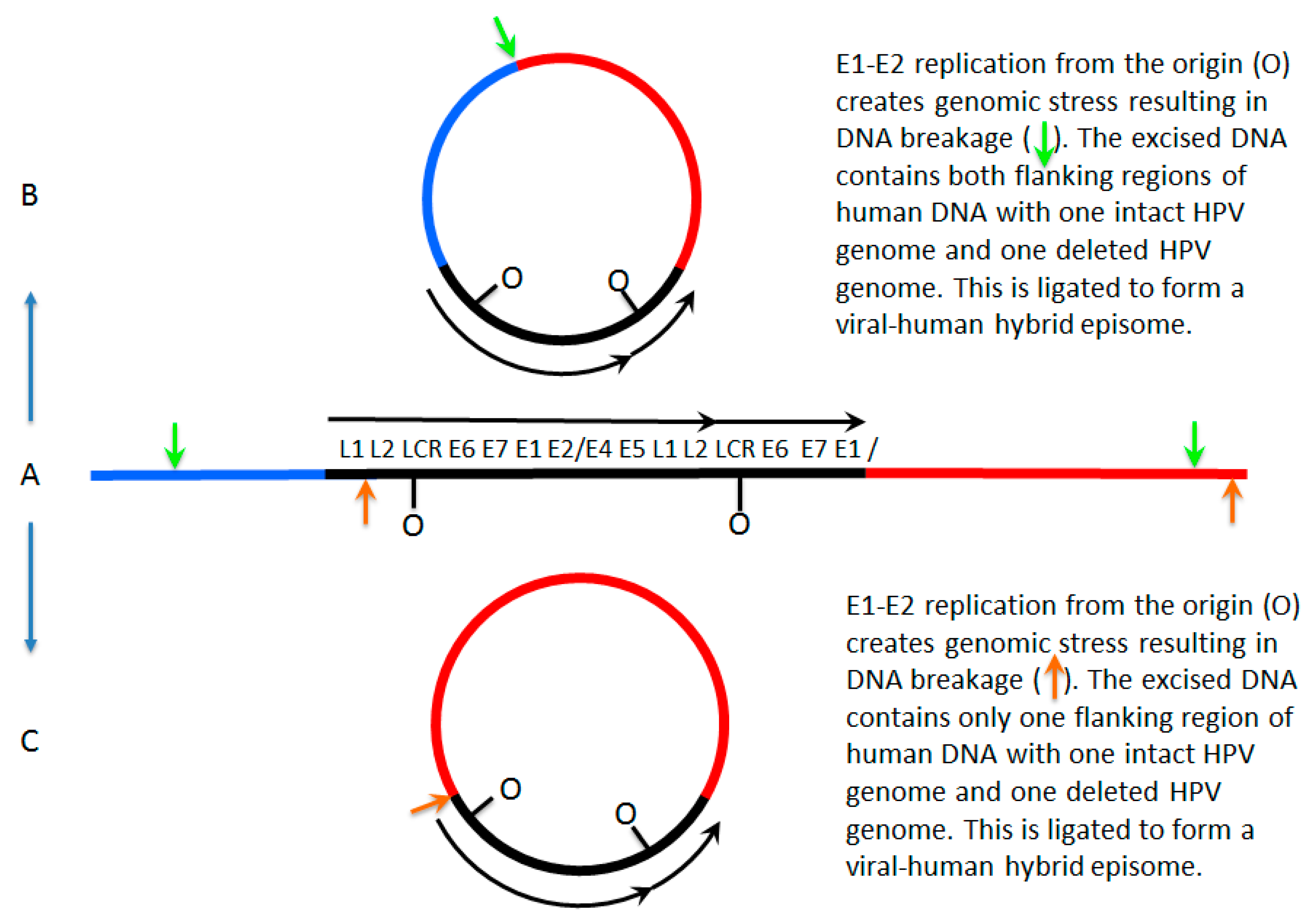
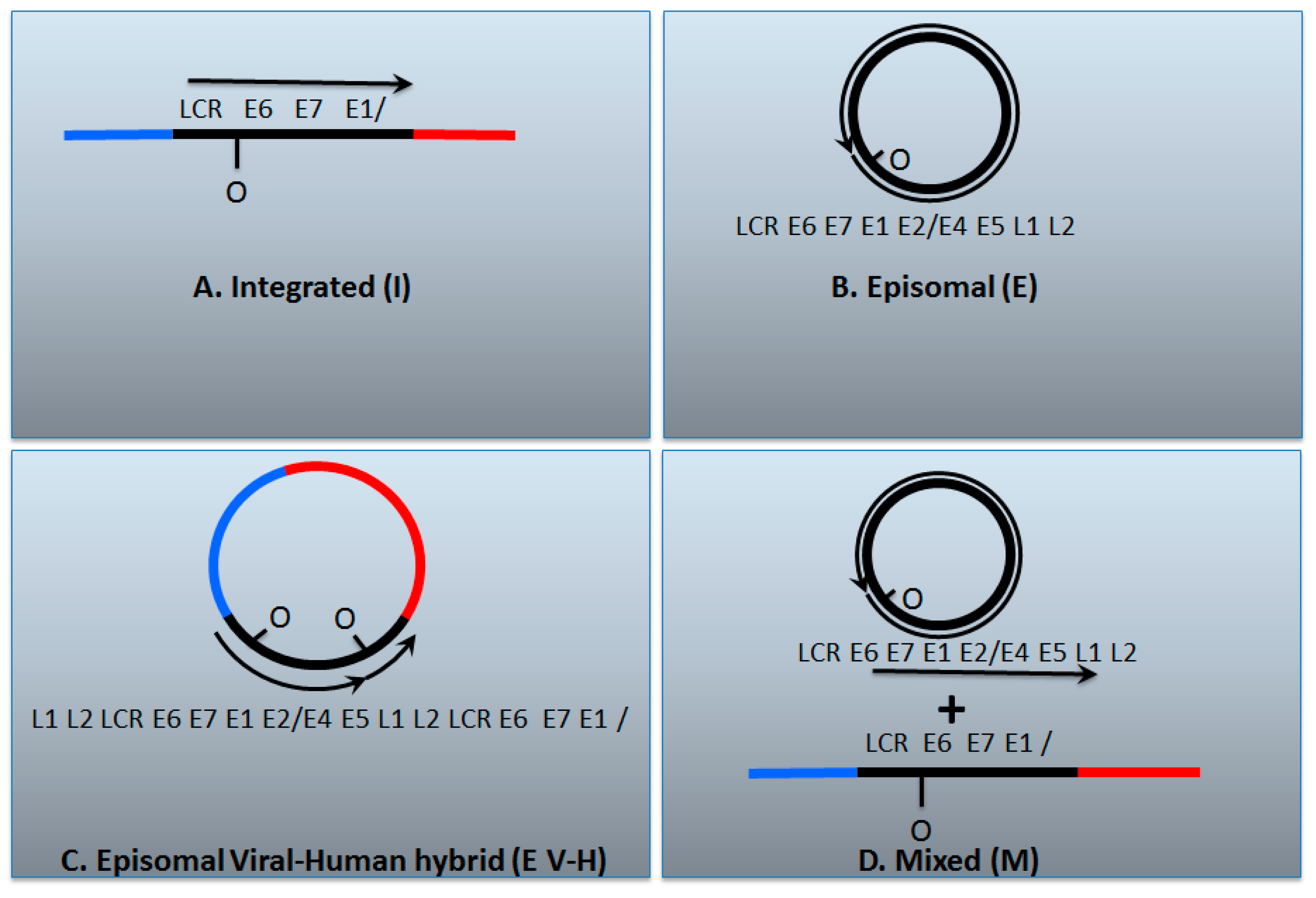
| I | E | E V-H | M | |
| E2/E6 DNA ratio is less than 1 | Y | N | Y | Y |
| Viral–human DNA hybrids | Y | N | Y | Y |
| Viral–human RNA hybrids | Y | N | Y | Y |
| Full length E2 RNA expression | N | Y | Y | Y |
| E5 RNA expression | N | Y | Y | Y |
| 8-kbp band on Southern blot following digestion with single cutter | N | Y | Y | Y |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgan, I.M.; DiNardo, L.J.; Windle, B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017, 9, 208. https://doi.org/10.3390/v9080208
Morgan IM, DiNardo LJ, Windle B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses. 2017; 9(8):208. https://doi.org/10.3390/v9080208
Chicago/Turabian StyleMorgan, Iain M., Laurence J. DiNardo, and Brad Windle. 2017. "Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift?" Viruses 9, no. 8: 208. https://doi.org/10.3390/v9080208
APA StyleMorgan, I. M., DiNardo, L. J., & Windle, B. (2017). Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses, 9(8), 208. https://doi.org/10.3390/v9080208