
Characterization of a Novel RNA Virus Discovered in the Autumnal Moth Epirrita autumnata in Sweden

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Origins and RNA Purification
2.2. Sequencing and Genome Assembly
2.3. Virus Quantification
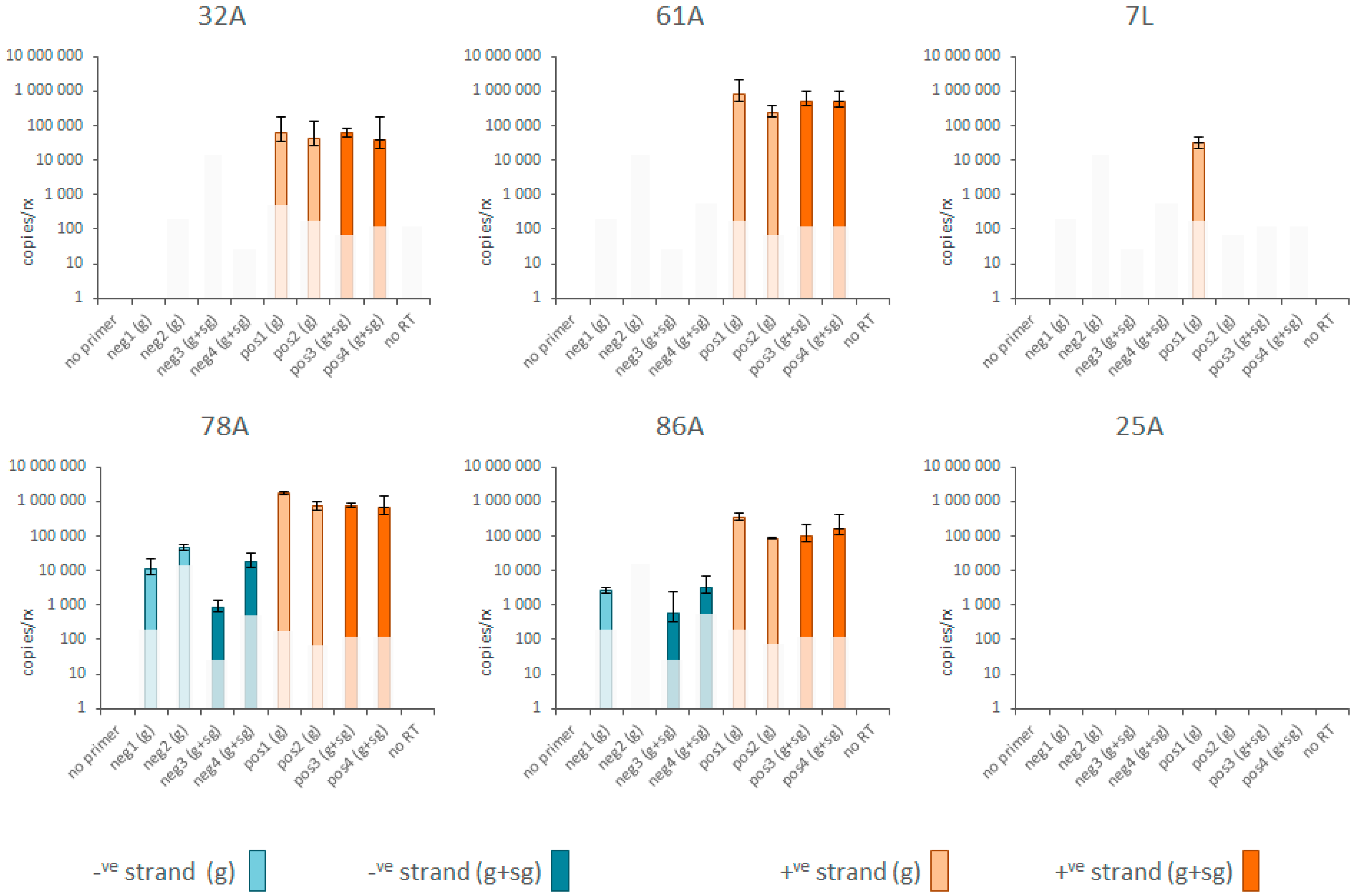
2.4. Positive and Negative Strand RNA Quantification
2.5. Phylogenetic Analyses
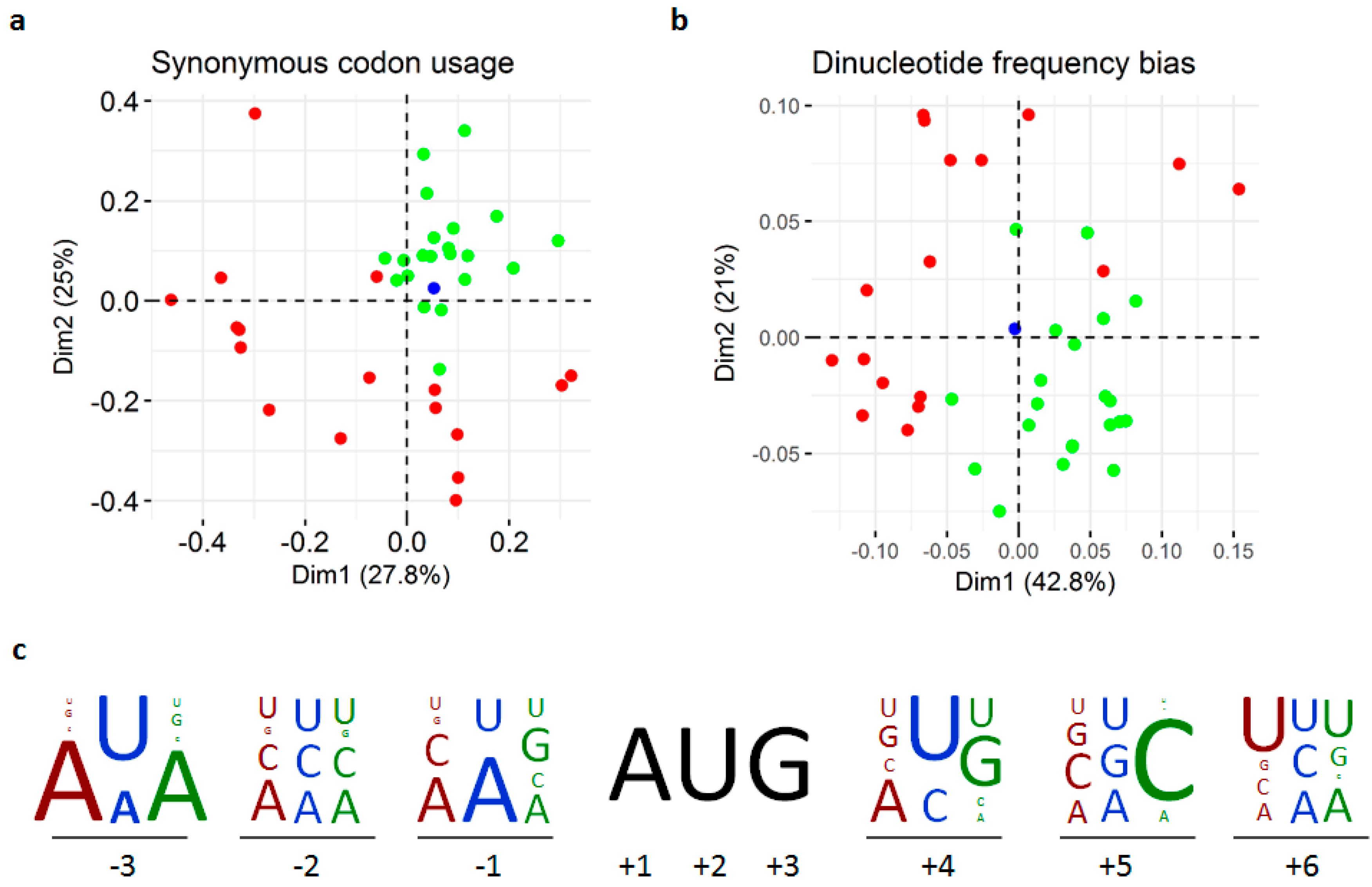
2.6. Compositional Bias Analyses
3. Results
3.1. Discovery
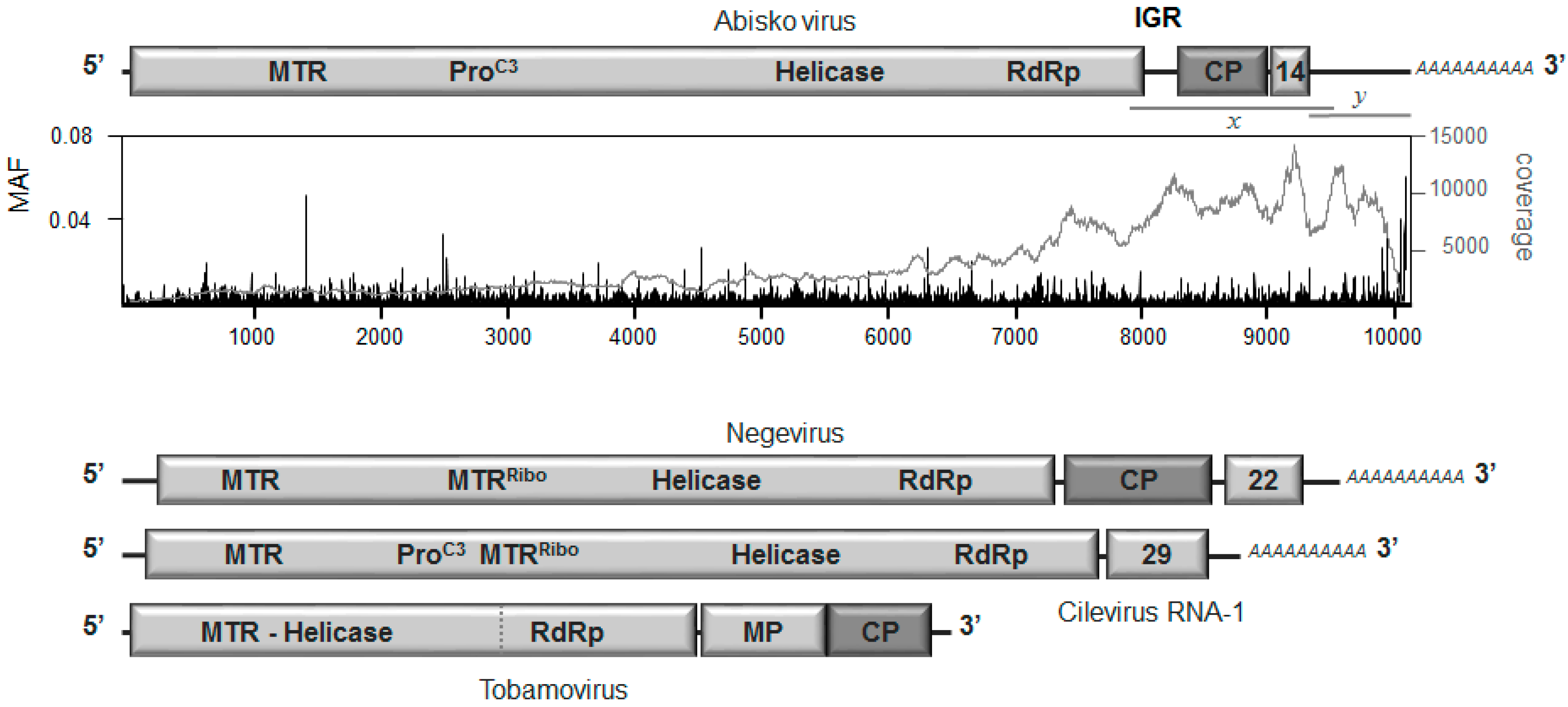
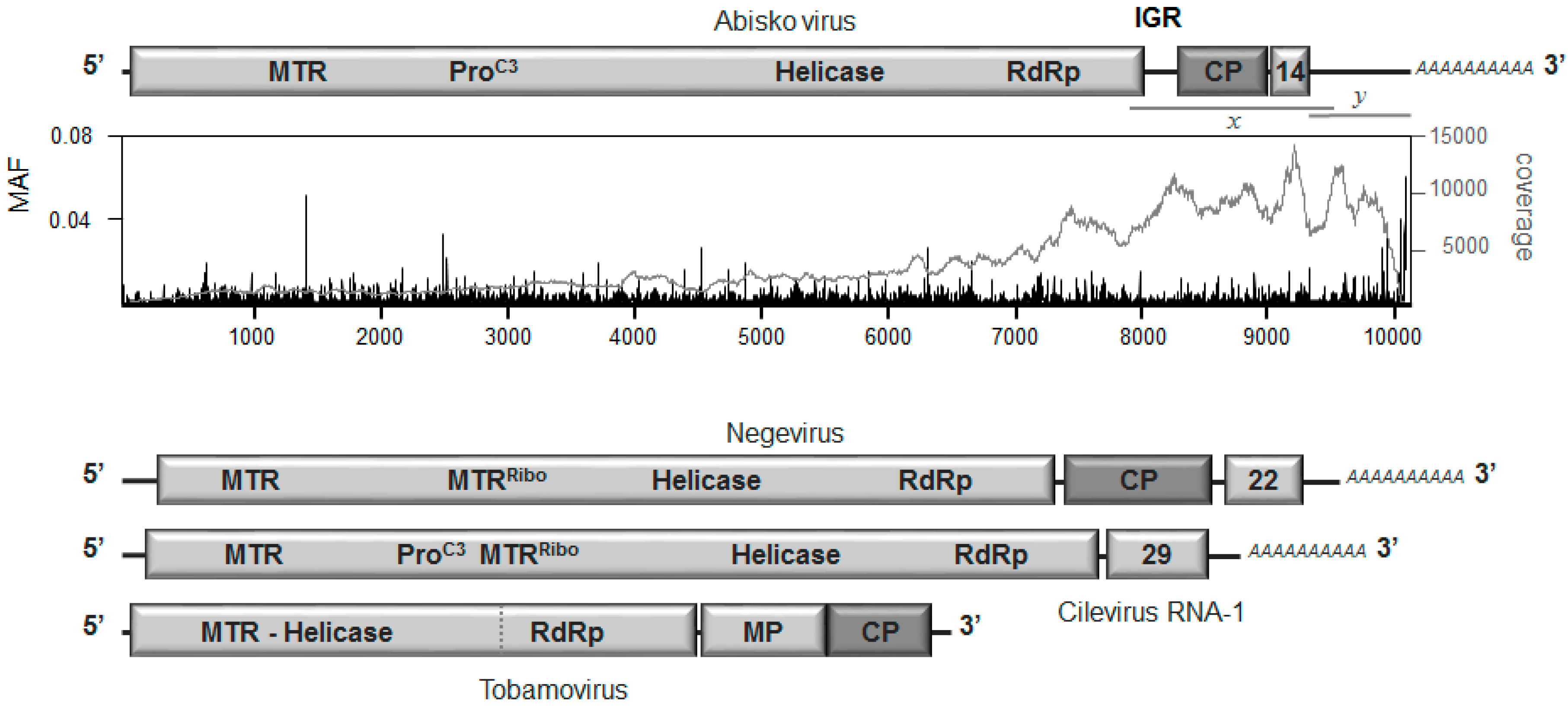
3.2. Genome Organization
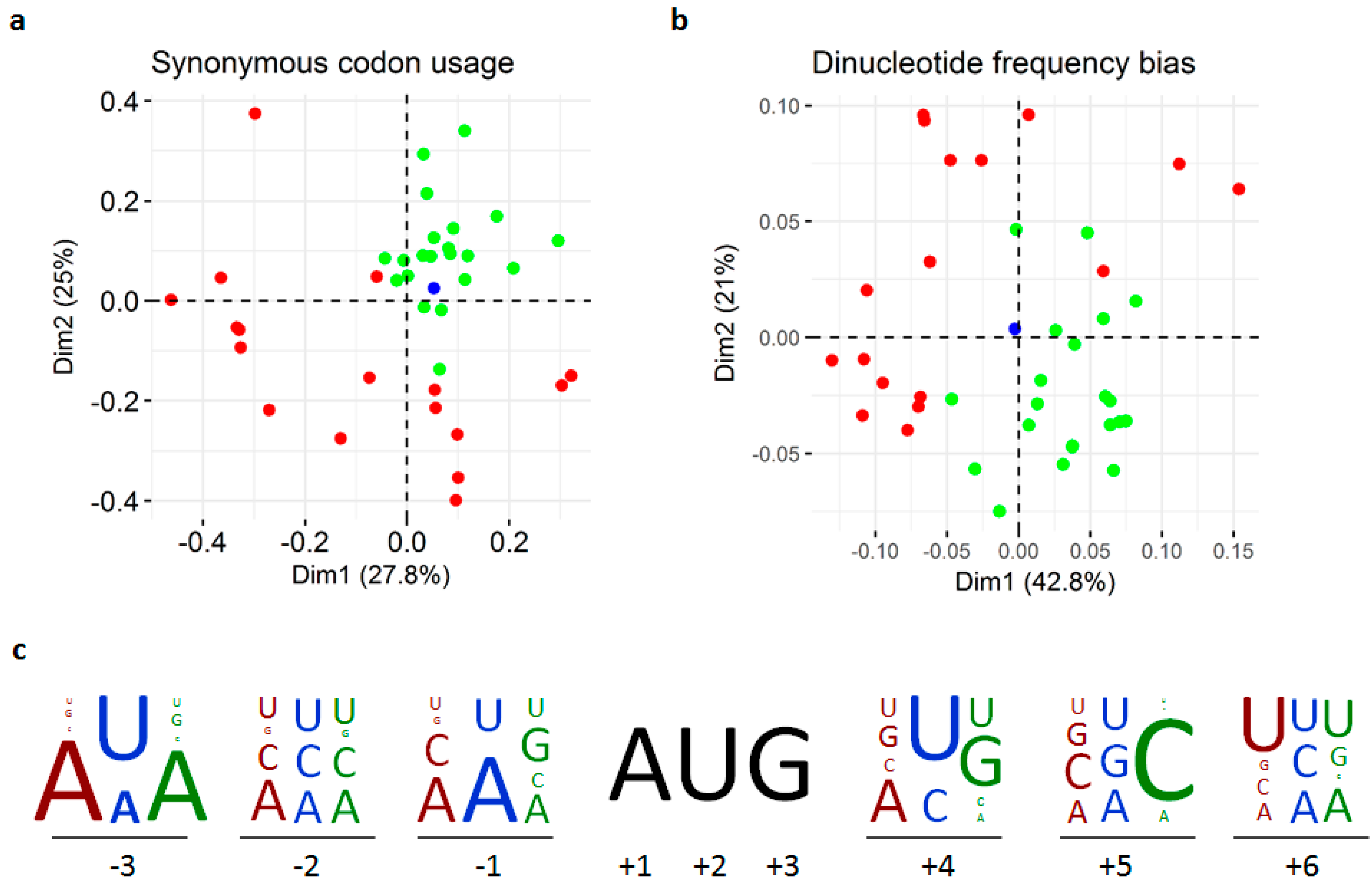
3.3. Replication and Translation
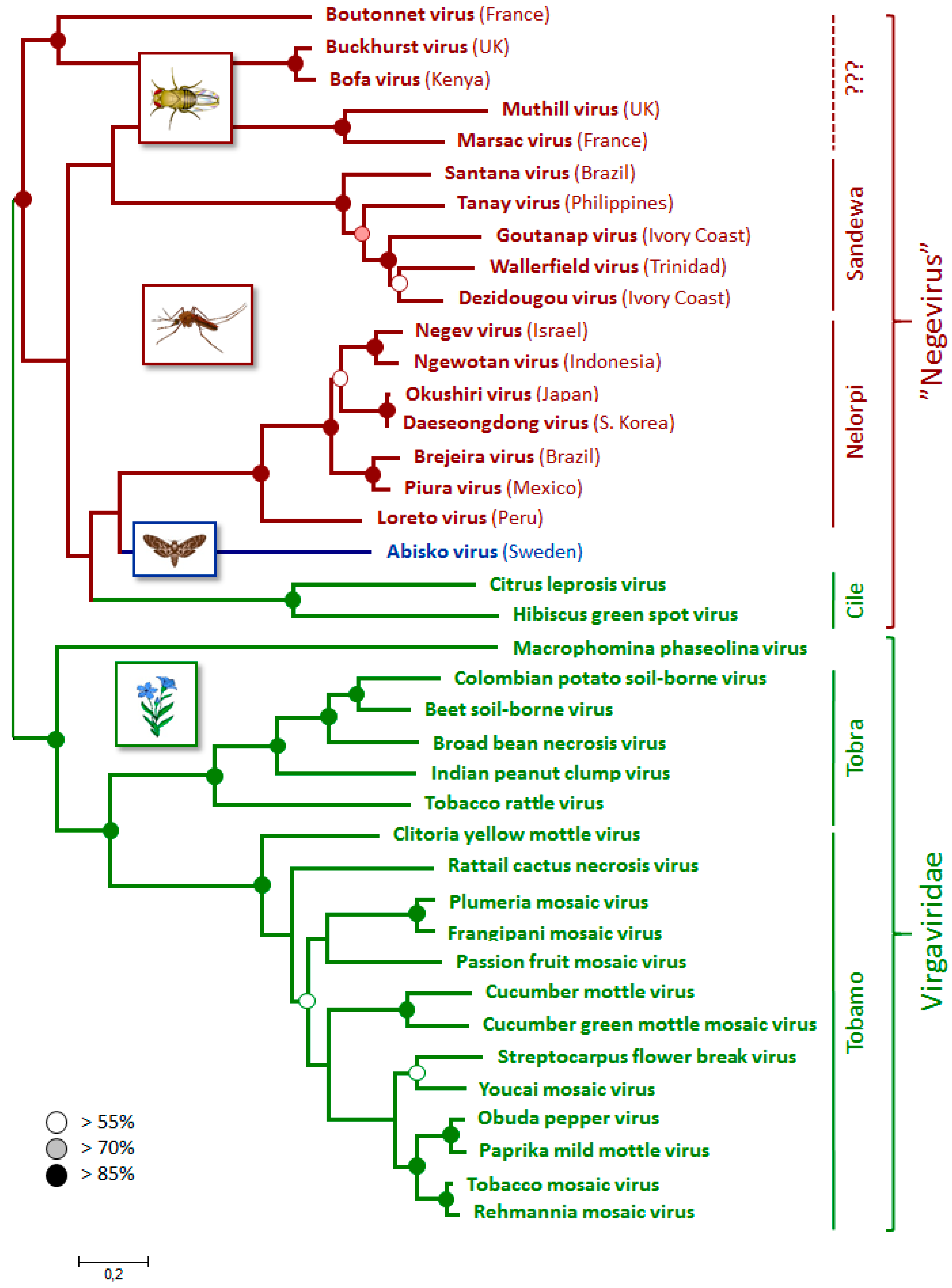
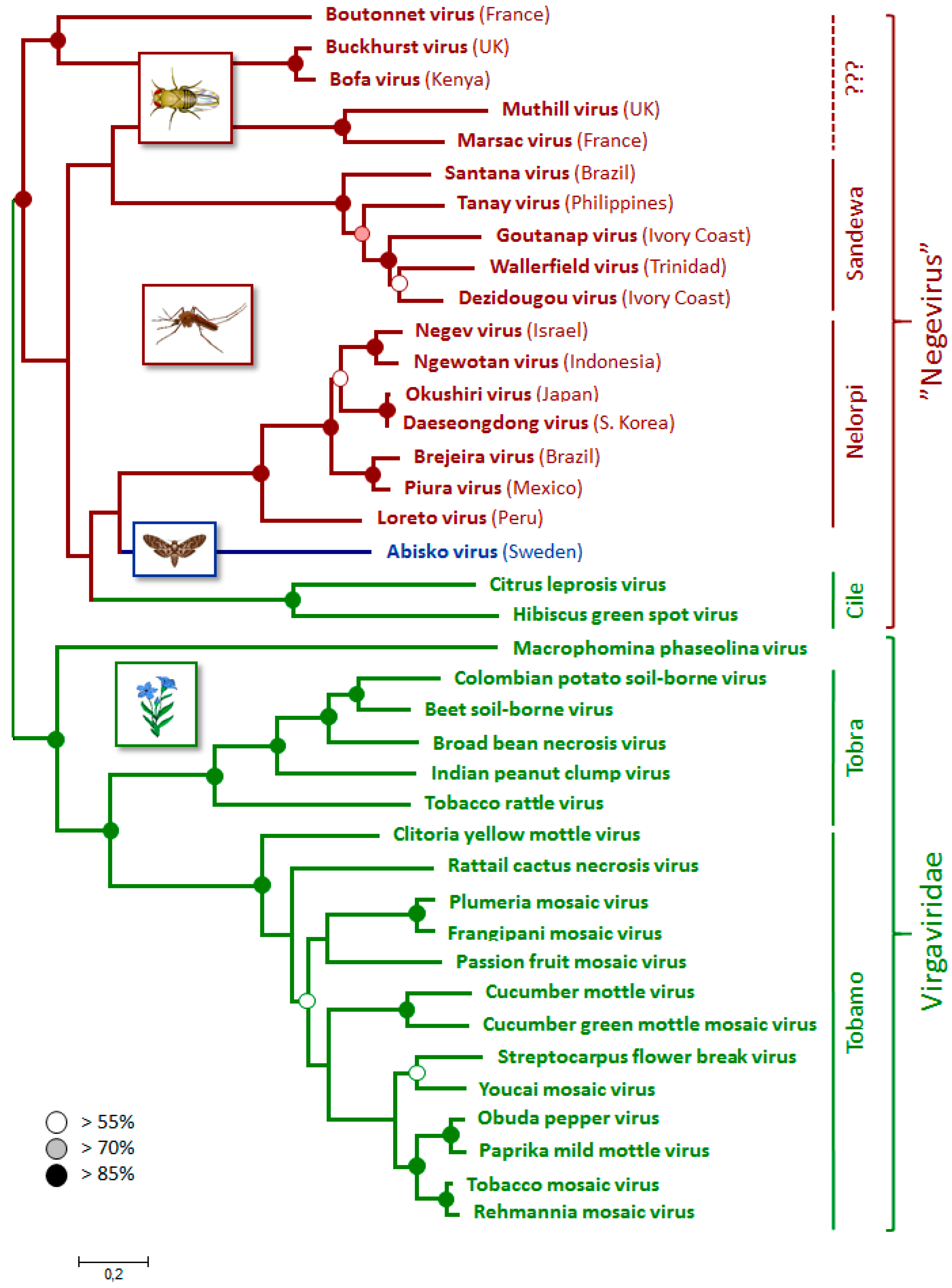
3.4. Phylogenetic Relationships
3.5. Incidence and Origin
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jepsen, J.U.; Hagen, S.B.; Ims, R.A.; Yoccoz, N.G. Climate change and outbreaks of the geometrids Operophtera brumata and Epirrita autumnata in subarctic birch forest: Evidence of a recent outbreak range expansion. J. Animal Ecol. 2008, 77, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Ammunét, T.; Bylund, H.; Jepsen, J.U. Northern Geometrids and Climate Change: From Abiotic Factors to Trophic Interactions. In Climate Change and Insect Pests; Björkman, C., Niemelä, P., Eds.; CABI: Wallingford, UK, 2015; pp. 235–247. [Google Scholar]
- Tenow, O.; Nilssen, A.C.; Bylund, H.; Hogstad, O. Waves and synchrony in Epirrita autumnata/Operophtera brumata outbreaks. I. Lagged synchrony: Regionally, locally and among species. J. Animal Ecol. 2007, 76, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Klemola, N.; Andersson, T.; Ruohomäki, K.; Klemola, T. Experimental test of parasitism hypothesis for population cycles of a forest lepidopteran. Ecology 2010, 9, 2506–2513. [Google Scholar] [CrossRef]
- Tenow, O. The outbreaks of Oporinia autumnata Bkh. and Operophtera spp. (Lep., Geometridae) in the Scandinavian mountain chain and northern Finland 1862–1968. Zool. Bidrag. Uppsalasuppl. 1972, 2, 1–107. [Google Scholar]
- Vasilakis, N.; Forrester, N.L.; Palacios, G.; Nasar, F.; Savji, N.; Rossi, S.L.; Guzman, H.; Wood, T.G.; Popov, V.; Gorchakov, R.; et al. Negevirus: A proposed new taxon of insect-specific viruses with wide geographic distribution. J. Virol. 2013, 87, 2475–2488. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.; Contreras-Guttierez, M.A.; Guzman, H.; Martins, L.C.; Barbirato, M.F.; Savit, C.; Balta, V.; Uribe, S.; Vivero, R.; Suaza, J.D.; et al. Genetic characterization, molecular epidemiology, and phylogenetic relationships of insect-specific viruses in the taxon Negevirus. Virology 2017, 504, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Nat. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Bailey, L.; Ball, B.V.; Blanchard, P.; Budge, G.; Chejanovsk, N.; Chen, Y.P.; Gauthier, L.; Genersch, E.; De Graaf, D.; et al. Standard methods for virus research in Apis mellifera. J. Apic. Res. 2013, 52, 1–56. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum information for publication of quantitative Real-Time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Craggs, J.K.; Ball, J.K.; Thomson, B.J.; Irving, W.L.; Grabowska, A.M. Development of a strand-specific RT-PCR based assay to detect the replicative form of hepatitis C virus RNA. J. Virol. Methods 2001, 94, 111–120. [Google Scholar] [CrossRef]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comp. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Bahir, I.; Fromer, M.; Linial, M. Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009, 5, e311. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Lipkin, W.I.; Zaidi, S.; Delwart, E. Use of nucleotide composition analysis to infer hosts for three novel picorna-like viruses. J. Virol. 2010, 84, 10322–10328. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [PubMed]
- Gu, W.; Zhou, T.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 2004, 101, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Dray, S.; Dufour, A.B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef]
- Jan, E. Divergent IRES elements in invertebrates. Virus Res. 2006, 119, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Joanna Sztuba-Solińska, J.; Stollar, V.; Bujarski, J.J. Subgenomic messenger RNAs: Mastering regulation of (+)-strand RNA virus life cycle. Virology 2011, 412, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Dreher, T.W.; Miller, W.A. Translational control in positive strand RNA plant viruses. Virology 2006, 344, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.A.; Karpova, O.V.; Skulachev, M.V.; Tomashevskaya, O.L.; Rodionova, N.P.; Dorokhov, Y.L.; Atabekov, J.G. A Tobamovirus genome that contains an internal ribosome entry site functional in vitro. Virology 1997, 232, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Ahlquist, P.; Noueiry, A.O.; Lee, W.M.; Kushner, D.B.; Dye, B.T. Host factors in positive-strand RNA virus genome replication. J. Virol. 2003, 77, 8181–8186. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.R.; Cornman, R.S.; Evans, J.D.; Semberg, E.; Haddad, N.; Neumann, P.; Gauthier, L. Genome characterization, prevalence and distribution of a Macula-like virus from Apis mellifera and Varroa destructor. Viruses 2015, 7, 3586–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, N.; Linial, M. Viral proteins acquired from a host converge to simplified domain architectures. PLoS Comput. Biol. 2012, 8, e1002364. [Google Scholar] [CrossRef] [PubMed]
- Tammaru, T.; Kaitaniemi, P.; Ruohomaki, K. Realized fecundity in Epirrita autumnata (Lepidoptera: Geometridae): Relation to body size and conse-quences to population dynamics. Oikos 1996, 77, 407–416. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Lat/Lon | Epirrita autumnata | Operophtera brumata | |||||
|---|---|---|---|---|---|---|---|---|
| La | Pu | Ad | La | Pu | Ad | |||
| Nuorgam | Finland | 70.09155, 27.89978 | 1 (0) | 9 (0) | - | - | 19 (0) | - |
| Utsjoki | Finland | 69.87178, 27.21863 | - | 6 (0) | - | - | - | - |
| Kilpisjärvi | Finland | 69.06390, 20.66048 | - | 18 (0) | 23 (1) | - | - | - |
| Neiden | Norway | 69.67379, 29.47426 | - | 12 (0) | - | - | 22 (0) | - |
| Gratangen | Norway | 68.72343, 17.59460 | - | 33 (0) | 24 (2) | - | 8 (0) | - |
| Abisko | Sweden | 68.30850, 18.51059 | 59 (1) | 8 (0) | 30 (1) | - | 3 (0) | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Miranda, J.R.; Hedman, H.; Onorati, P.; Stephan, J.; Karlberg, O.; Bylund, H.; Terenius, O. Characterization of a Novel RNA Virus Discovered in the Autumnal Moth Epirrita autumnata in Sweden. Viruses 2017, 9, 214. https://doi.org/10.3390/v9080214
De Miranda JR, Hedman H, Onorati P, Stephan J, Karlberg O, Bylund H, Terenius O. Characterization of a Novel RNA Virus Discovered in the Autumnal Moth Epirrita autumnata in Sweden. Viruses. 2017; 9(8):214. https://doi.org/10.3390/v9080214
Chicago/Turabian StyleDe Miranda, Joachim R., Harald Hedman, Piero Onorati, Jörg Stephan, Olof Karlberg, Helena Bylund, and Olle Terenius. 2017. "Characterization of a Novel RNA Virus Discovered in the Autumnal Moth Epirrita autumnata in Sweden" Viruses 9, no. 8: 214. https://doi.org/10.3390/v9080214
APA StyleDe Miranda, J. R., Hedman, H., Onorati, P., Stephan, J., Karlberg, O., Bylund, H., & Terenius, O. (2017). Characterization of a Novel RNA Virus Discovered in the Autumnal Moth Epirrita autumnata in Sweden. Viruses, 9(8), 214. https://doi.org/10.3390/v9080214