
Deciphering Single Nucleotide Polymorphisms and Evolutionary Trends in Isolates of the Cydia pomonella granulovirus

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. CpGV Isolates and Sequences
2.2. Genome Sequencing
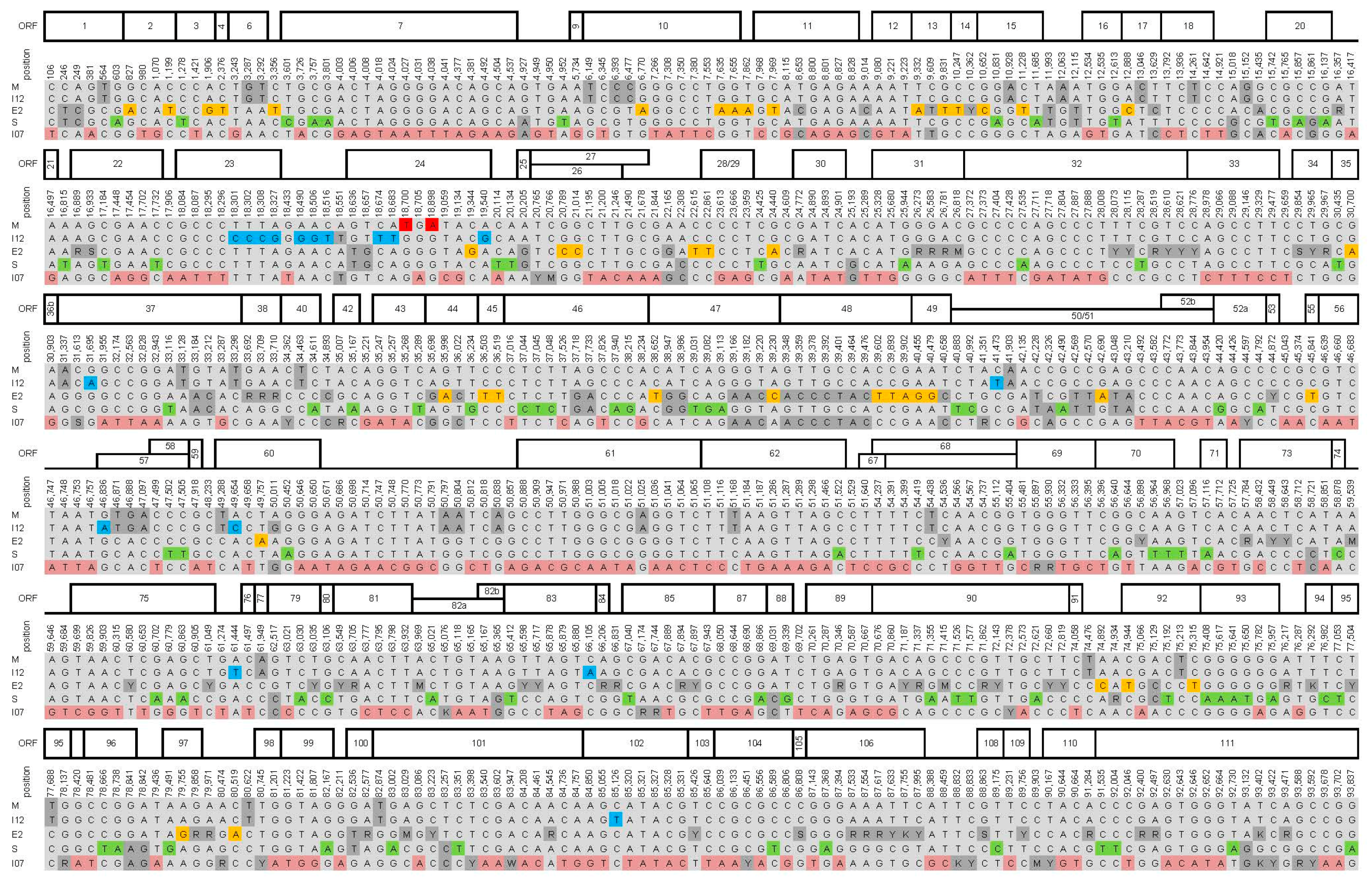
2.3. Genome Analysis
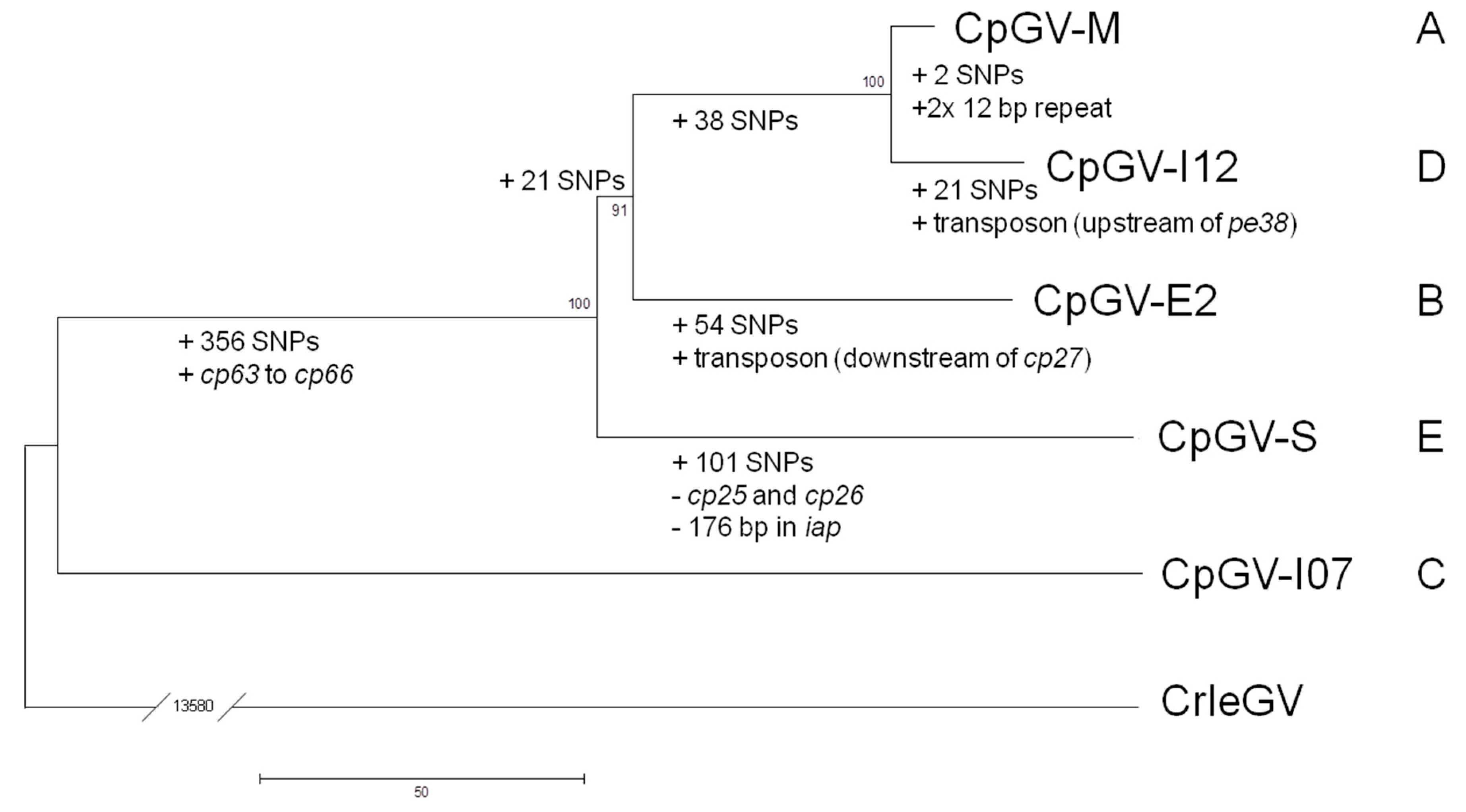
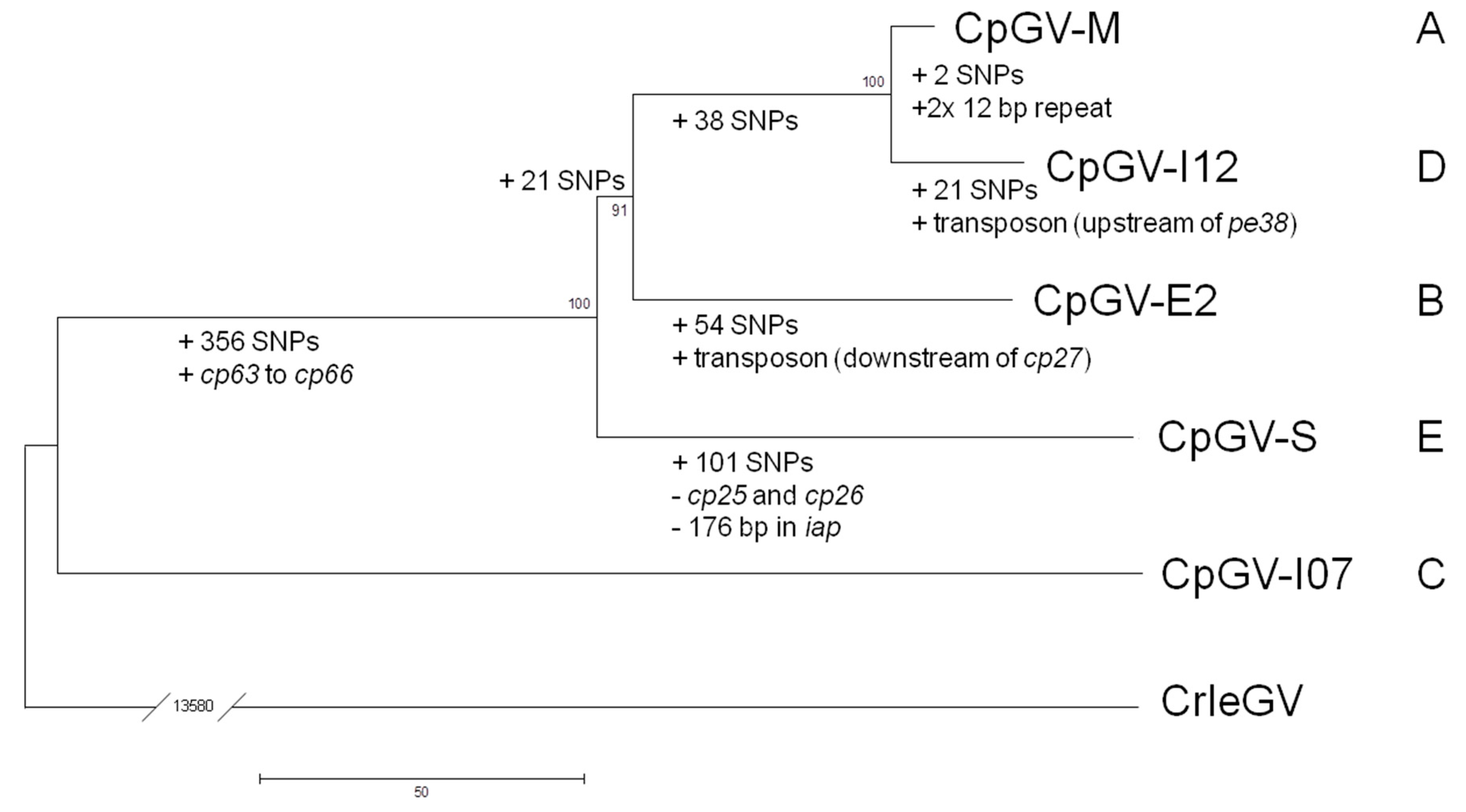
2.4. Phylogenetic Analysis
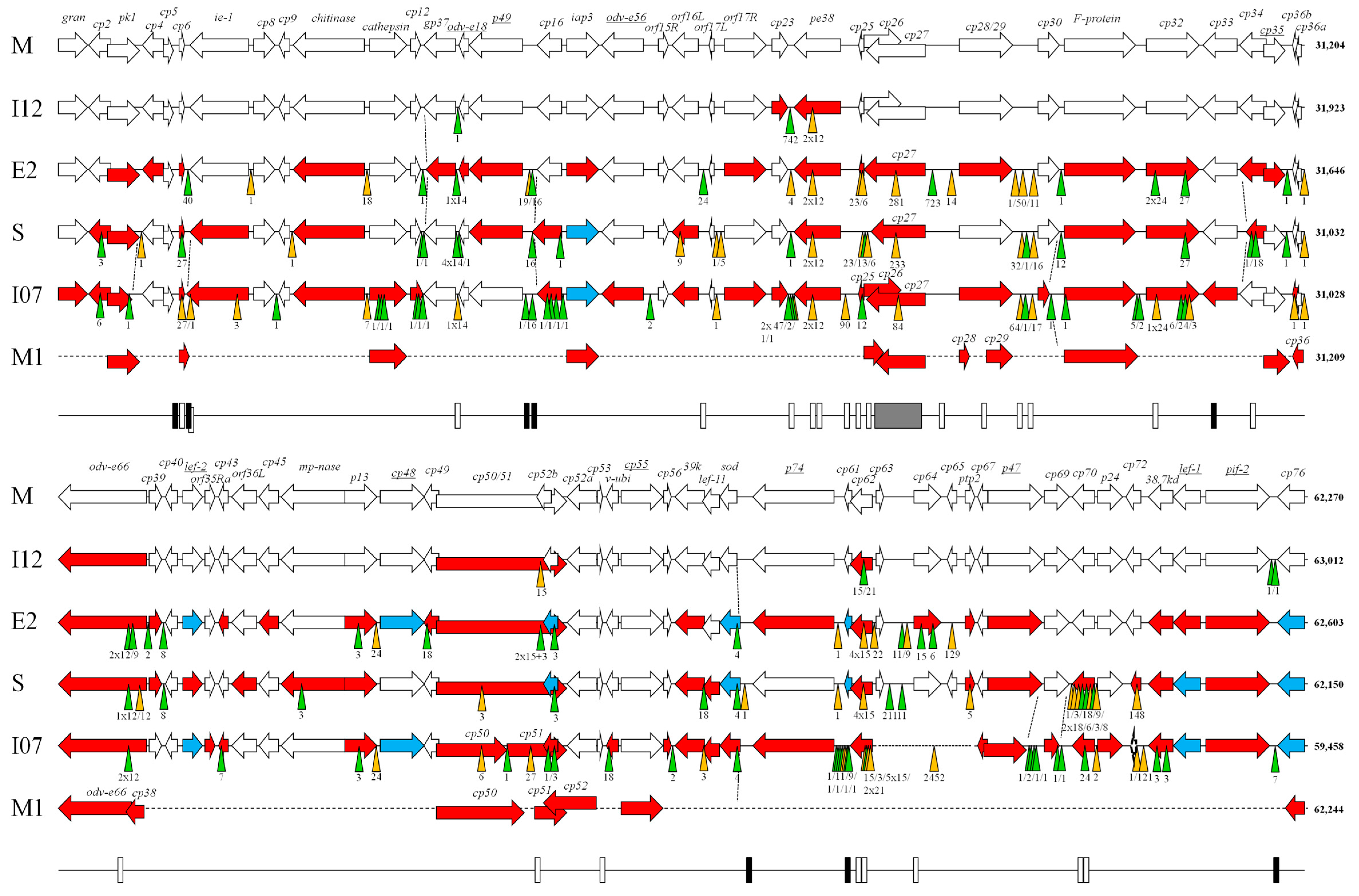
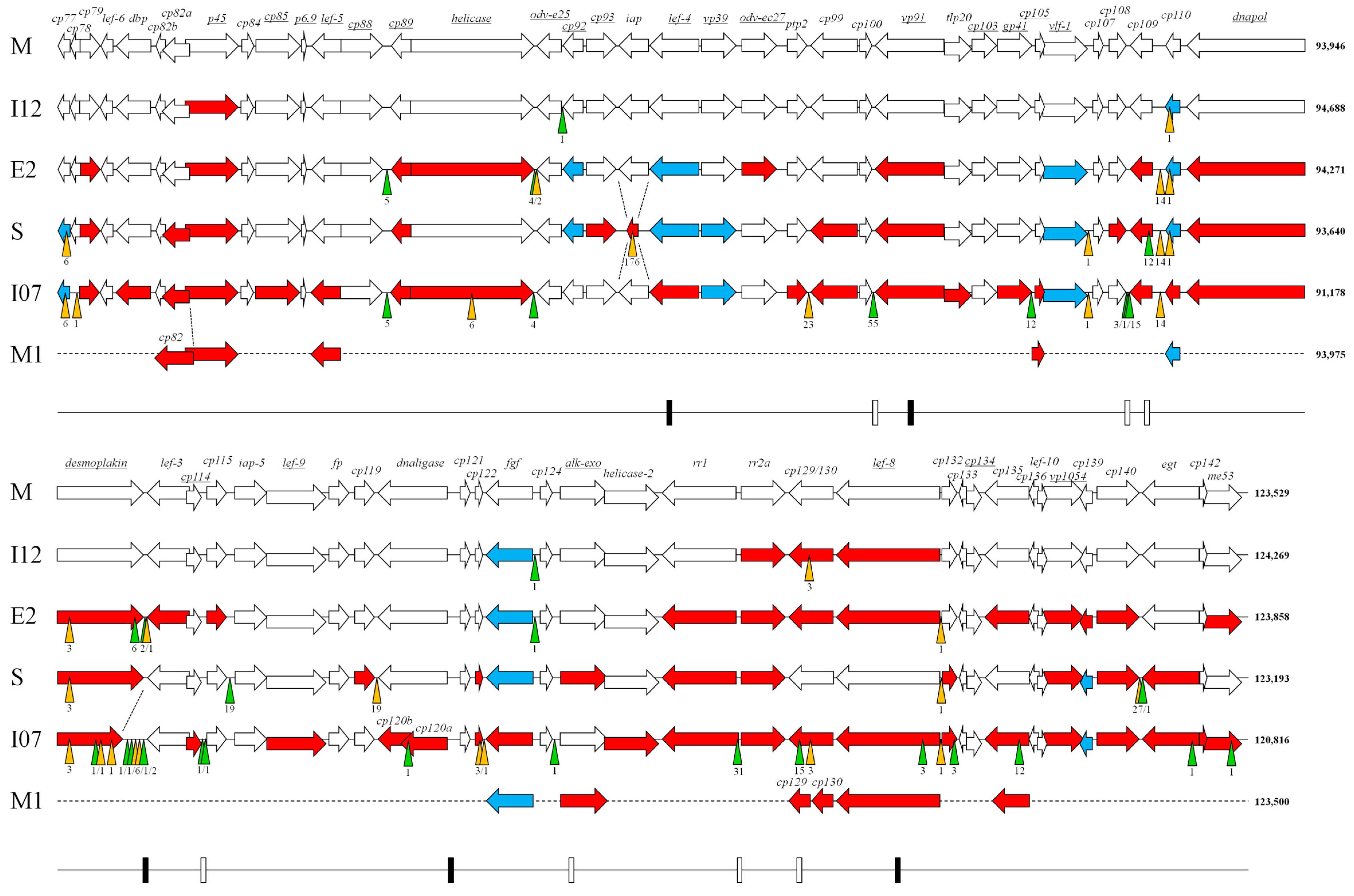
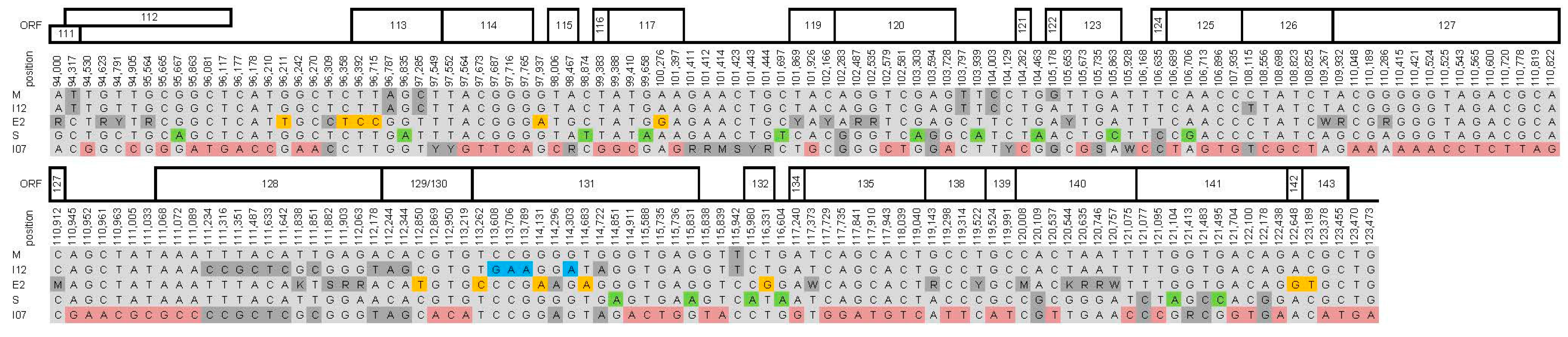
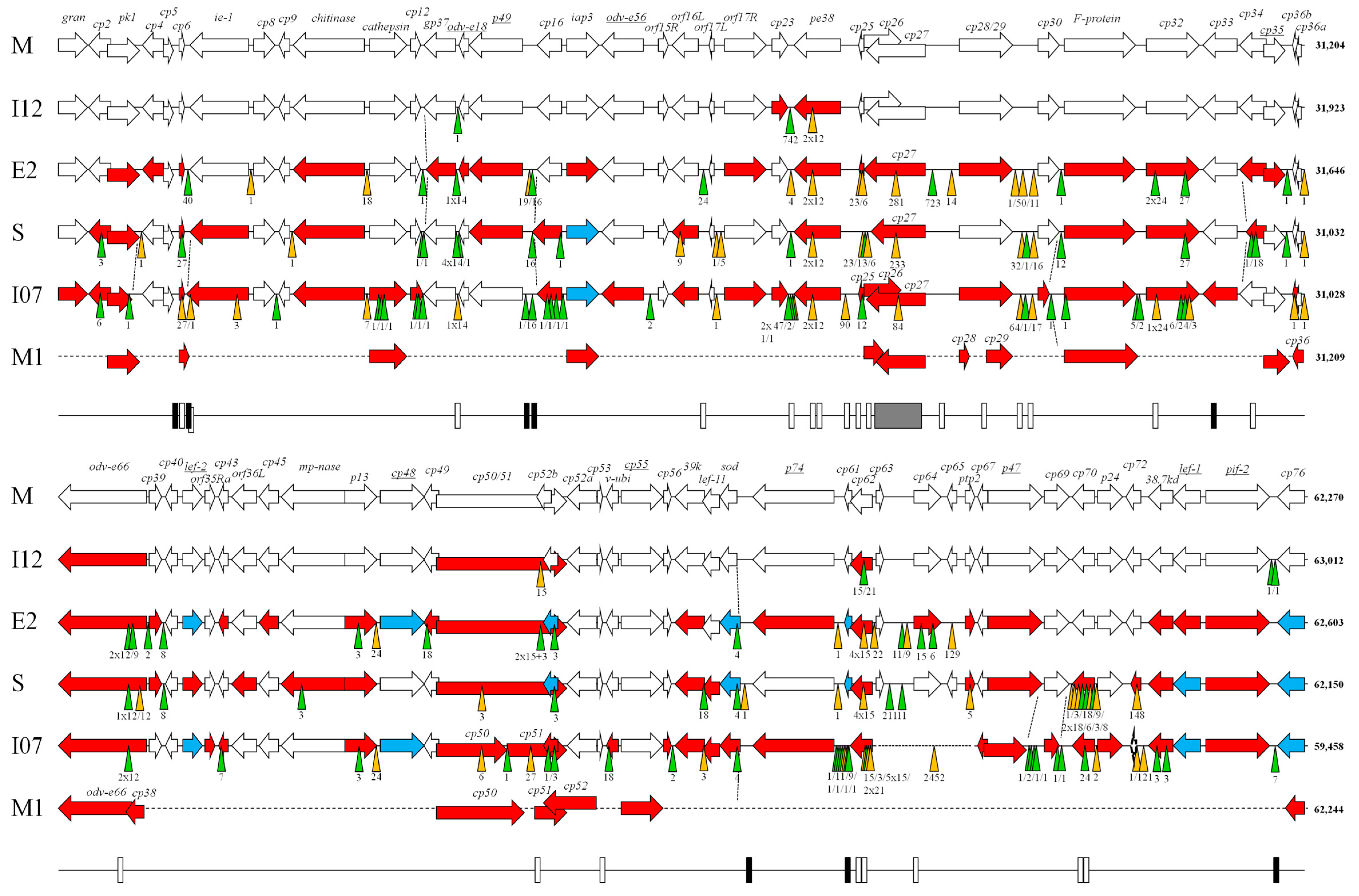
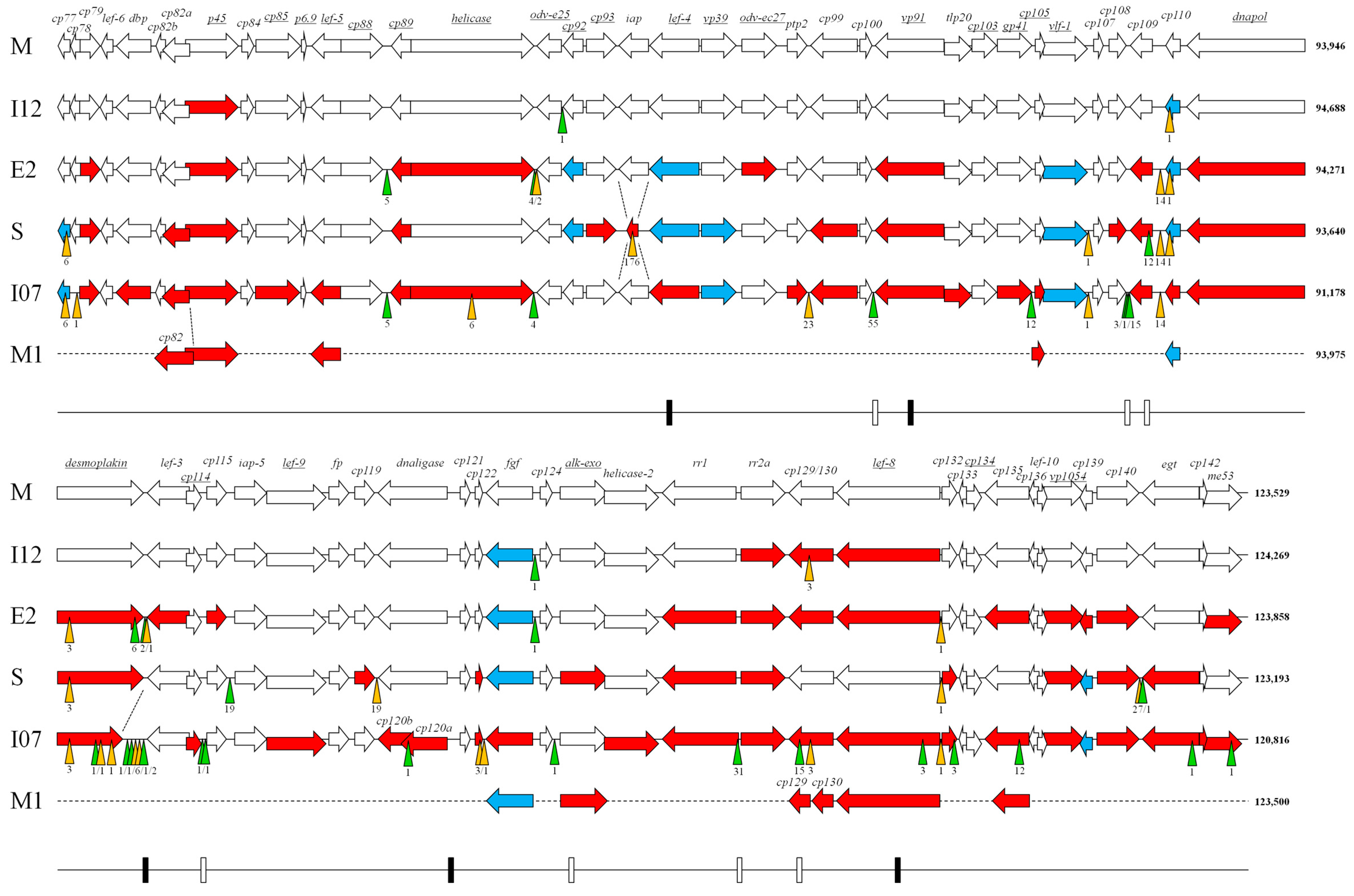
3. Results and Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jehle, J.A.; Blissard, G.W.; Bonning, B.C.; Cory, J.S.; Herniou, E.A.; Rohrmann, G.F.; Theilmann, D.A.; Thiem, S.M.; Vlak, J.M. On the classification and nomenclature of baculoviruses: A proposal for revision. Arch. Virol. 2006, 151, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Arif, B.M.; Becnel, J.J.; Blissard, G.W.; Bonning, B.C.; Harrison, R.L.; Jehle, J.A.; Theilmann, D.A.; Vlak, J.M. Baculoviridae. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Oxford, UK, 2011; pp. 163–174. [Google Scholar]
- Eberle, K.E.; Jehle, J.A.; Huber, J. Integrated pest management: Principles and practice. In Microbial Control of Crop Pests Using Insect Viruses; Abrol, D.P., Shanker, U., Eds.; CABI: Wallingford, UK, 2012; pp. 281–298. [Google Scholar]
- Cross, J.V.; Solomon, M.G.; Chandler, D.; Jarrett, P.; Richardson, P.N.; Winstanley, D.; Bathon, H.; Huber, J.; Keller, B.; Langenbruch, G.A.; et al. Biocontrol of pests of apples and pears in northern and central Europe: 1. Microbial agents and nematodes. Biocontrol Sci. Technol. 1999, 9, 125–149. [Google Scholar] [CrossRef]
- Gebhardt, M.M.; Eberle, K.E.; Radtke, P.; Jehle, J.A. Baculovirus resistance in codling moth is virus isolate-dependent and the consequence of a mutation in viral gene pe38. Proc. Natl. Acad. Sci. USA 2014, 111, 15711–15716. [Google Scholar] [CrossRef] [PubMed]
- Eberle, K.E.; Asser-Kaiser, S.; Sayed, S.M.; Nguyen, H.T.; Jehle, J.A. Overcoming the resistance of codling moth against conventional Cydia pomonella granulovirus (CpGV-M) by a new isolate CpGV-I12. J. Invertebr. Pathol. 2008, 98, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Eberle, K.E.; Sayed, S.; Rezapanah, M.; Shojai-Estabragh, S.; Jehle, J.A. Diversity and evolution of the Cydia pomonella granulovirus. J. Gen. Virol. 2009, 90, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Luque, T.; Finch, R.; Crook, N.; O’Reilly, D.R.; Winstanley, D. The complete sequence of the Cydia pomonella granulovirus genome. J. Gen. Virol. 2001, 82, 2531–2547. [Google Scholar] [CrossRef] [PubMed]
- Tanada, Y. A granulosis virus of the codling moth, Carpocapsa pomonella (Linnaeus) (Olethreutidae, Lepidoptera). J. Invertebr. Pathol. 1964, 6, 378–380. [Google Scholar]
- Crook, N.E.; Spencer, R.A.; Payne, C.C.; Leisy, D.J. Variation in Cydia pomonella granulosis virus isolates and physical maps of the DNA from three variants. J. Gen. Virol. 1985, 2423–2430. [Google Scholar] [CrossRef]
- Rezapanah, M.; Shojai-Estabragh, S.; Huber, J.; Jehle, J.A. Molecular and biological characterization of new isolates of Cydia pomonella granulovirus from Iran. J. Pest Sci. 2008, 81, 187–191. [Google Scholar] [CrossRef]
- Huber, J. Western Europe. In Insect Viruses and Pest Management; Hunter-Fujita, F.R., Entwistle, P.F., Evans, H.F., Crook, N.E., Eds.; Wiley: New York, NY, USA, 1998; pp. 201–215. [Google Scholar]
- Asser-Kaiser, S.; Fritsch, E.; Undorf-Spahn, K.; Kienzle, J.; Eberle, K.E.; Gund, N.A.; Reineke, A.; Zebitz, C.P.; Heckel, D.G.; Huber, J.; et al. Rapid emergence of baculovirus resistance in codling moth due to dominant, sex-linked inheritance. Science 2007, 317, 1916–1918. [Google Scholar] [CrossRef] [PubMed]
- Berling, M.; Blachere-Lopez, C.; Soubabere, O.; Lery, X.; Bonhomme, A.; Sauphanor, B.; Lopez-Ferber, M. Cydia pomonella granulovirus Genotypes Overcome Virus Resistance in the Codling Moth and Improve Virus Efficiency by Selection against Resistant Hosts. Appl. Environ. Microbiol. 2009, 75, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Asser-Kaiser, S.; Radtke, P.; El-Salamouny, S.; Winstanley, D.; Jehle, J.A. Baculovirus resistance in codling moth (Cydia pomonella L.) caused by early block of virus replication. Virology 2011, 410, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Zichová, T.; Falta, V.; Kocourek, F.; Stará, J. Others Differences in the susceptibility of codling moth populations to Cydia pomonella granulovirus in the Czech Republic. HortScience 2011, 38, 21–26. [Google Scholar]
- Jehle, J.A.; Schulze-Bopp, S.; Undorf-Spahn, K.; Fritsch, E. Evidence for a second type of resistance against Cydia pomonella granulovirus in field populations of codling moths. Appl. Environ. Microbiol. 2017, 83, e02330-16. [Google Scholar] [CrossRef] [PubMed]
- Zingg, D. Madex Plus and Madex I12 overcome virus resistance of codling moth. In Proceedings of the 13th International Conference on Cultivation Technique and Phytopathological Problems in Organic Fruit-Growing, Weinsberg, Germany, 18–20 February 2008; pp. 256–260. [Google Scholar]
- Carstens, E.B. Introduction to Virus Taxonomy. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Oxford, UK, 2011; pp. 3–7. [Google Scholar]
- Van Regenmortel, M.H. Virus species, a much overlooked but essential concept in virus classification. Intervirology 1990, 31, 241–254. [Google Scholar] [CrossRef]
- Brito, A.F.; Braconi, C.T.; Weidmann, M.; Dilcher, M.; Alves, J.M.; Gruber, A.; Zanotto, P.M. The Pangenome of the Anticarsia gemmatalis Multiple Nucleopolyhedrovirus (AgMNPV). Genome Biol. Evol. 2016, 8, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Craveiro, S.R.; Santos, L.A.V.M.; Togawa, R.C.; Inglis, P.W.; Grynberg, P.; Ribeiro, Z.M.A.; Ribeiro, B.M.; Castro, M.E.B. Complete genome sequences of six Chrysodeixis includens nucleopolyhedrovirus isolates from Brazil and Guatemala. Genome Announc. 2016, 4, e01192-16. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.P.; Cheng, R.L.; Xi, Y.; Zhang, C.X. Genomic diversity of Bombyx mori nucleopolyhedrovirus strains. Genomics 2013, 102, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucl. Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Carstens, E.B.; Wu, Y. No single homologous repeat region is essential for DNA replication of the baculovirus Autographa californica multiple nucleopolyhedrovirus. J. Gen. Virol. 2007, 88, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; van den Berg, P.M.; Tramper, J.; Goldbach, R.W.; Vlak, J.M. Location of two putative origins of DNA repliation of Autographa californica nuclear polyhedrosis virus. Virology 1993, 192, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.N.; Rohrmann, G.F. Lymantria dispar nuclear polyhedrosis virus homologous regions: Characterization of their ability to function as replication origins. J. Virol. 1995, 69, 213–221. [Google Scholar] [PubMed]
- Lange, M.; Jehle, J.A. The genome of the Cryptophlebia leucotreta granulovirus. Virology 2003, 317, 220–236. [Google Scholar] [CrossRef]
- Jehle, J.A.; Fritsch, E.; Nickel, E.; Huber, J.; Backhaus, H. TCl4.7: A novel lepidopteran transposon found in Cydia pomonella granulosis virus. Virology 1995, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; van der Linden, I.F.; Backhaus, H.; Vlak, J.M. Identification and sequence analysis of the integration site of transposon TCp3.2 in the genome of Cydia pomonella granulovirus. Virus Res. 1997, 50, 151–157. [Google Scholar] [PubMed]
- Arends, H.M.; Jehle, J.A. Homologous recombination between the inverted terminal repeats of defective transposon TCp3. 2 causes an inversion in the genome of Cydia pomonella granulovirus. J. Gen. Virol. 2002, 83, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Peccoud, J.; Chateigner, A.; Moumen, B.; Cordaux, R.; Herniou, E.A. Continuous influx of genetic material from host to virus populations. PLoS Genet. 2016, 12, e1005838. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
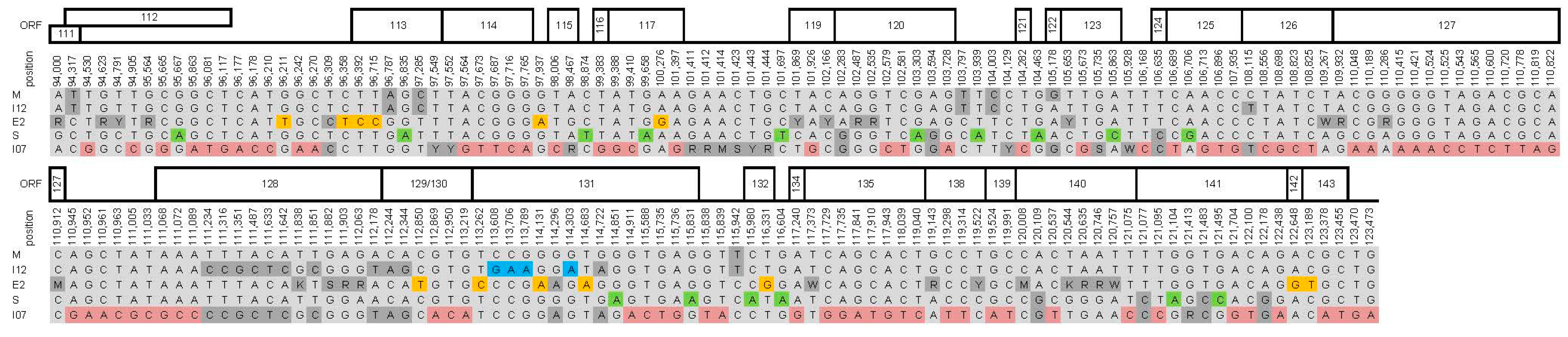{kind=link}
| Genome Feature | CpGV Isolate | ||||
|---|---|---|---|---|---|
| M | I12 | E2 | S | I07 | |
| sequencing method | 454 | Sanger | 454 | 454 | 454 |
| average coverage | 17.23 | 3.9 | 243 | 22.21 | 8.05 |
| genome length (bp) | 123,529 | 124,269 | 123,858 | 123,193 | 120,816 |
| coding (bp) | 113,121 | 113,145 | 111,822 | 111,372 | 109,635 |
| (% of total size) | (91.57) | (91.05) | (90.28) | (90.40) | (90.75) |
| non-coding (bp) | 10,408 | 11,124 | 12,036 | 11,821 | 11,181 |
| (% of total size) | (8.43) | (8.95) | (9.72) | (9.60) | (9.25) |
| GC% | 45.27 | 45.21 | 45.21 | 45.28 | 45.39 |
| number of open reading frames (ORF) | 142 | 142 | 140 | 140 | 137 |
| ORFs with identical amino acid (aa) compared to CpGV-M | 137 | 77 | 76 | 48 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wennmann, J.T.; Radtke, P.; Eberle, K.E.; Gueli Alletti, G.; Jehle, J.A. Deciphering Single Nucleotide Polymorphisms and Evolutionary Trends in Isolates of the Cydia pomonella granulovirus. Viruses 2017, 9, 227. https://doi.org/10.3390/v9080227
Wennmann JT, Radtke P, Eberle KE, Gueli Alletti G, Jehle JA. Deciphering Single Nucleotide Polymorphisms and Evolutionary Trends in Isolates of the Cydia pomonella granulovirus. Viruses. 2017; 9(8):227. https://doi.org/10.3390/v9080227
Chicago/Turabian StyleWennmann, Jörg T., Pit Radtke, Karolin E. Eberle, Gianpiero Gueli Alletti, and Johannes A. Jehle. 2017. "Deciphering Single Nucleotide Polymorphisms and Evolutionary Trends in Isolates of the Cydia pomonella granulovirus" Viruses 9, no. 8: 227. https://doi.org/10.3390/v9080227
APA StyleWennmann, J. T., Radtke, P., Eberle, K. E., Gueli Alletti, G., & Jehle, J. A. (2017). Deciphering Single Nucleotide Polymorphisms and Evolutionary Trends in Isolates of the Cydia pomonella granulovirus. Viruses, 9(8), 227. https://doi.org/10.3390/v9080227