
Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
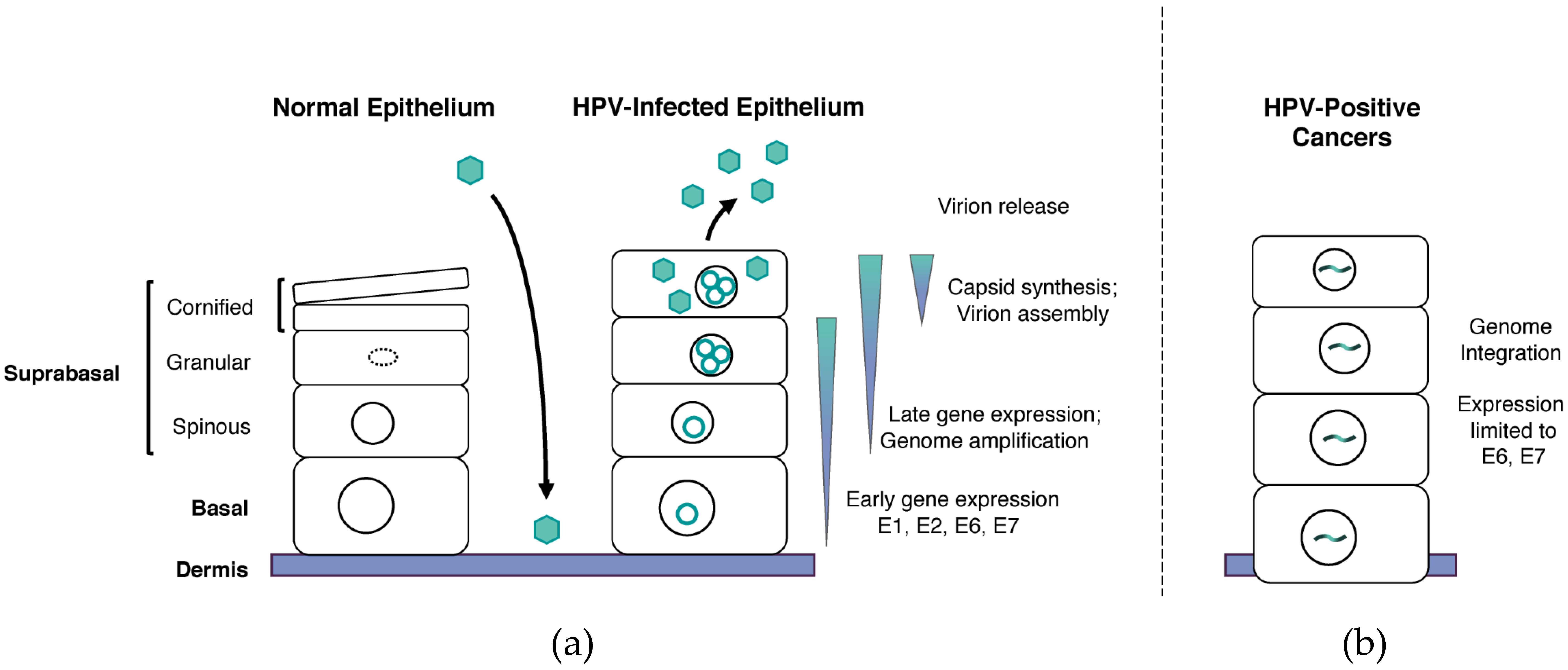
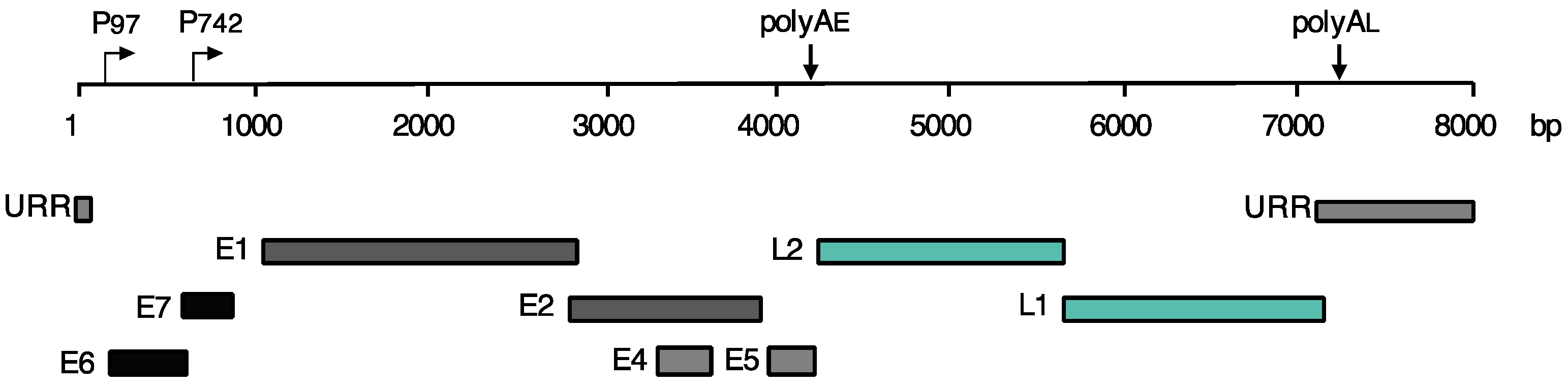
2. Life Cycle of HPVs
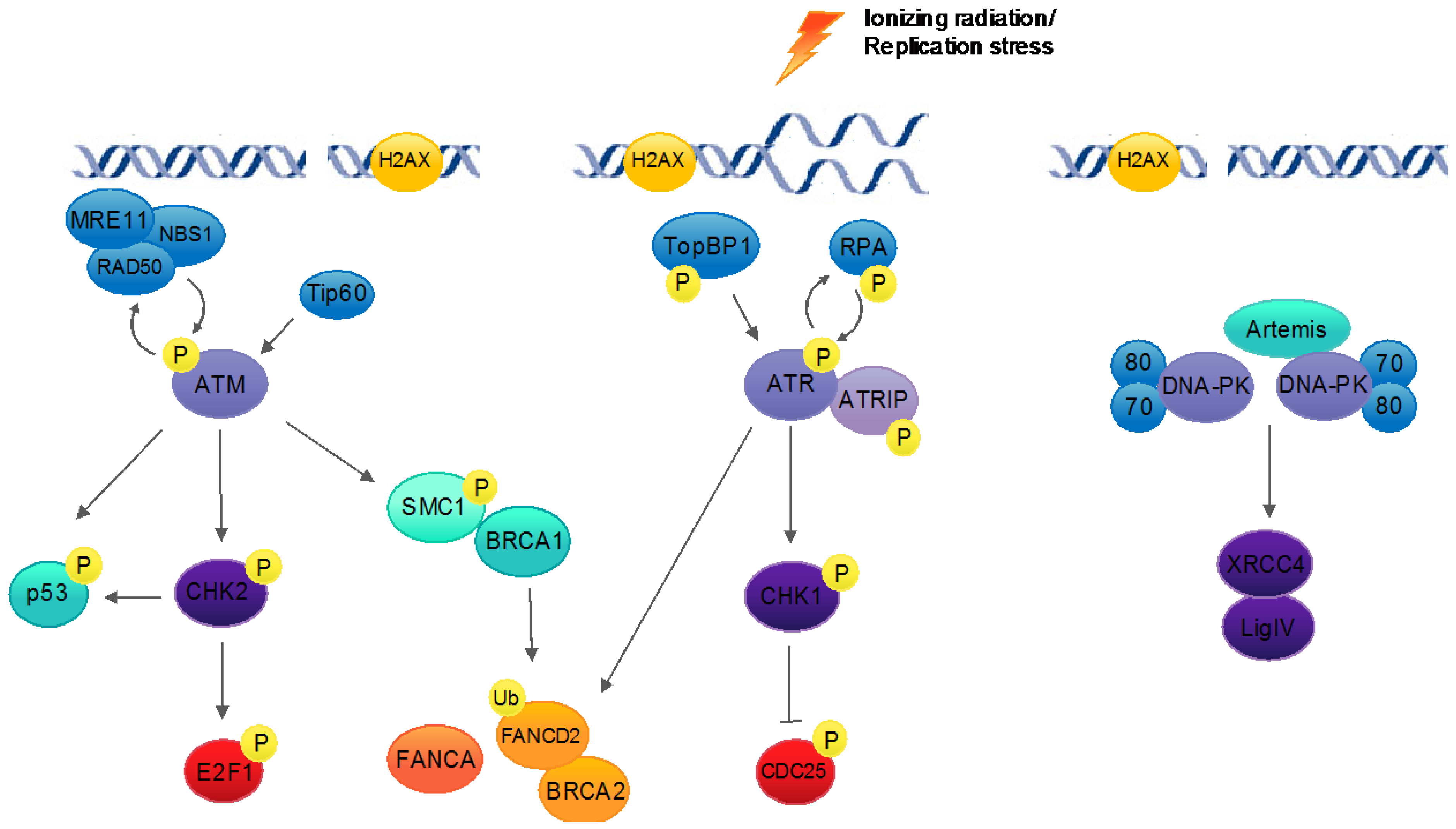
3. The Host DNA Damage Response
4. HPV Manipulation of the DNA Damage Response
4.1. ATM Pathway Activation Is Necessary for Productive Replication
4.2. ATR Pathway Activation Is Required for Viral Replication
4.3. Inhibition of Downstream Signaling Pathways by E6 and E7
4.4. The FA Pathway in Viral Replication and Transformation
5. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; Zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Munger, K. The role of human papillomaviruses in human cancers. Front. Biosci. 2002, 7, d641–d649. [Google Scholar] [CrossRef] [PubMed]
- Viens, L.J.; Henley, S.J.; Watson, M.; Markowitz, L.E.; Thomas, C.C.; Thompson, T.D.; Razzaghi, H.; Saraiya, M. Human papillomavirus-associated cancers-United States, 2008–2012. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.R.; McCrory, D.C.; Nanda, K.; Bastian, L.; Matchar, D.B. Mathematical model for the natural history of human papillomavirus infection and cervical carcinogenesis. Am. J. Epidemiol. 2000, 151, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Saraiya, M.; Steben, M.; Watson, M.; Markowitz, L. Evolution of cervical cancer screening and prevention in United States and Canada: Implications for public health practitioners and clinicians. Prev. Med. 2013, 57, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.C., Jr.; Massad, L.S.; Dunton, C.J.; Spitzer, M.; Wilkinson, E.J.; Solomon, D.; American Society for Colposcopy; Cervical Pathology-sponsored Consensus Conference. 2006 consensus guidelines for the management of women with cervical intraepithelial neoplasia or adenocarcinoma in situ. Am. J. Obstet. Gynecol. 2007, 197, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E. Epidermal differentiation and keratin gene expression. Princess Takamatsu Symp. 1994, 24, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Laimins, L.A. Human papillomavirus life cycle: Active and latent phases. Semin. Cancer Biol. 1999, 9, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F. Papillomavirus DNA replication. J. Virol. 1991, 65, 3417–3420. [Google Scholar] [PubMed]
- Cheng, S.; Schmidt-Grimminger, D.C.; Murant, T.; Broker, T.R.; Chow, L.T. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995, 9, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Laimins, L.A. The differentiation-dependent life cycle of human papillomaviruses in keratinocytes. In The Papillomaviruses; Dimaio, D., Garcea, D., Eds.; Springer: New York, NY, USA, 2007; pp. 45–68. [Google Scholar]
- Wang, H.K.; Duffy, A.A.; Broker, T.R.; Chow, L.T. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev. 2009, 23, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Rohlfs, M.; Winkenbach, S.; Meyer, S.; Rupp, T.; Durst, M. Viral transcription in human keratinocyte cell lines immortalized by human papillomavirus type-16. Virology 1991, 183, 331–342. [Google Scholar] [CrossRef]
- Del Vecchio, A.M.; Romanczuk, H.; Howley, P.M.; Baker, C.C. Transient replication of human papillomavirus DNAs. J. Virol. 1992, 66, 5949–5958. [Google Scholar] [PubMed]
- Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002, 76, 11291–11300. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [PubMed]
- Wilson, R.; Ryan, G.B.; Knight, G.L.; Laimins, L.A.; Roberts, S. The full-length E1^E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology 2007, 362, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, F.; Klumpp, D.J.; Laimins, L.A. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 2003, 77, 2819–2831. [Google Scholar] [CrossRef] [PubMed]
- Genther, S.M.; Sterling, S.; Duensing, S.; Munger, K.; Sattler, C.; Lambert, P.F. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 2003, 77, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Frattini, M.G.; Laimins, L.A. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein. Proc. Natl Acad. Sci. USA 1994, 91, 12398–12402. [Google Scholar] [CrossRef] [PubMed]
- Sedman, J.; Stenlund, A. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J. 1995, 14, 6218–6228. [Google Scholar] [PubMed]
- Mohr, I.J.; Clark, R.; Sun, S.; Androphy, E.J.; MacPherson, P.; Botchan, M.R. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 1990, 250, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Eyfjord, J.E.; Bodvarsdottir, S.K. Genomic instability and cancer: Networks involved in response to DNA damage. Mutat. Res. 2005, 592, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sulli, G.; Di Micco, R.; d’Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed]
- Grompe, M. FANCD2: A branch-point in DNA damage response? Nat. Med. 2002, 8, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi Anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Garcia-Higuera, I.; Xu, B.; Andreassen, P.R.; Gregory, R.C.; Kim, S.T.; Lane, W.S.; Kastan, M.B.; D’Andrea, A.D. Convergence of the Fanconi Anemia and ataxia telangiectasia signaling pathways. Cell 2002, 109, 459–472. [Google Scholar] [CrossRef]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; McNamara, G.; Cheng, K.T.; Huang, J.; Wang, C.H.; Comai, L.; Ou, J.H.; Lai, M.M. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J. Immunol. 2010, 185, 6985–6998. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.R.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and Tip60 histone acetyltransferase to promote virus replication. Cell Host Microbe 2011, 10, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.C.; Hussmann, K.L.; McBride, A.A. The role of the DNA damage response throughout the papillomavirus life cycle. Viruses 2015, 7, 2450–2469. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Vande Pol, S.; Banerjee, N.S.; Dutta, A.B.; Chow, L.T.; Dutta, A. Destabilization of Tip60 by human papillomavirus E6 results in attenuation of Tip60-dependent transcriptional regulation and apoptotic pathway. Mol. Cell 2010, 38, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dutta, A.; Laimins, L.A. The acetyltransferase Tip60 is a critical regulator of the differentiation-dependent amplification of human papillomaviruses. J. Virol. 2015, 89, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor NBS1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Gautam, D.; Ok, S.T.; Johnson, B.A.; Anacker, D.C.; Moody, C.A. Homologous recombination repair factors Rad51 and BRCA1 are necessary for productive replication of human papillomavirus 31. J. Virol. 2015, 90, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- Langsfeld, E.S.; Bodily, J.M.; Laimins, L.A. The deacetylase sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and Rad51 to viral genomes. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar] [PubMed]
- Sowd, G.A.; Mody, D.; Eggold, J.; Cortez, D.; Friedman, K.L.; Fanning, E. SV40 utilizes ATM kinase activity to prevent non-homologous end joining of broken viral DNA replication products. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Gunasekharan, V.; Satsuka, A.; Laimins, L.A. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Nishino, T.; Marko, J.F. The SMC1–SMC3 cohesin heterodimer structures DNA through supercoiling-dependent loop formation. Nucleic Acids Res. 2013, 41, 6149–6160. [Google Scholar] [CrossRef] [PubMed]
- Feeney, K.M.; Wasson, C.W.; Parish, J.L. Cohesin: A regulator of genome integrity and gene expression. Biochem. J. 2010, 428, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 regulates transcription of the topoisomerase II beta-binding protein 1 (TopBP1) gene to activate the ATR pathway and promote human papillomavirus replication. Am. Soc. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [PubMed]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Gunawardena, R.W.; Habash, K.B.; Wise-Draper, T.M.; Jansen, M.; Knudsen, E.S.; Wells, S.I. Coordinate regulation of Fanconi Anemia gene expression occurs through the RB/E2F pathway. Oncogene 2008, 27, 4798–4808. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Thompson, D.A.; Munger, K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 1997, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [PubMed]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Wreesmann, V.B.; Goberdhan, A.; Ben-Porat, L.; Satagopan, J.; Ngai, I.; Huvos, A.G.; Giampietro, P.; Levran, O.; Pujara, K.; et al. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi Anemia patients. J. Natl. Cancer Inst. 2003, 95, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R.; Gillison, M.L. A new link between Fanconi Anemia and human papillomavirus-associated malignancies. J. Natl. Cancer Inst. 2003, 95, 1648–1650. [Google Scholar] [CrossRef] [PubMed]
- Spardy, N.; Duensing, A.; Charles, D.; Haines, N.; Nakahara, T.; Lambert, P.F.; Duensing, S. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi Anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J. Virol. 2007, 81, 13265–13270. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morreale, R.J.; Werner, S.P.; Higginbotham, J.M.; Laimins, L.A.; Lambert, P.F.; Brown, D.R.; Gillison, M.L.; Nuovo, G.J.; Witte, D.P.; et al. The Fanconi Anemia pathway limits human papillomavirus replication. J. Virol. 2012, 86, 8131–8138. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morris, T.A.; Higginbotham, J.M.; Spardy, N.; Cha, E.; Kelly, P.; Williams, D.A.; Wikenheiser-Brokamp, K.A.; Duensing, S.; Wells, S.I. Fanconi Anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene 2009, 28, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Pitot, H.C.; Strati, K.; Spardy, N.; Duensing, S.; Grompe, M.; Lambert, P.F. Deficiencies in the Fanconi Anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res. 2010, 70, 9959–9968. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Laimins, L.A. FANCD2 binds human papillomavirus genomes and associates with a distinct set of DNA repair proteins to regulate viral replication. Microbiology 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Karttunen, H.; Savas, J.N.; McKinney, C.; Chen, Y.H.; Yates, J.R., 3rd; Hukkanen, V.; Huang, T.T.; Mohr, I. Co-opting the Fanconi Anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth. Mol. Cell 2014, 55, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, G.; Naim, V.; Caruso, P.; Burla, R.; Bogliolo, M.; Cundari, E.; Benihoud, K.; Saggio, I.; Rosselli, F. The FANC pathway is activated by adenovirus infection and promotes viral replication-dependent recombination. Nucleic Acids Res. 2011, 39, 5459–5473. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Hu, L.; Hein, J.; Gjoerup, O.V. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large t antigen. J. Virol. 2010, 84, 8007–8020. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Garcia-Higuera, I.; Andreassen, P.R.; Gregory, R.C.; Grompe, M.; D’Andrea, A.D. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and Rad51. Blood 2002, 100, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Andreassen, P.R.; D’Andrea, A.D. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol. Cell. Biol. 2004, 24, 5850–5862. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; de Vries, Y.; Rooimans, M.A.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi Anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2007, 39, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; D’Andrea, A.D. How the Fanconi Anemia pathway guards the genome. Ann. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Hoskins, E.E.; Privette Vinnedge, L.M.; Foglesong, G.D.; Brusadelli, M.G.; Potter, S.S.; Komurov, K.; Brugmann, S.A.; Lambert, P.F.; Kimple, R.J.; et al. Defects in the Fanconi Anemia pathway in head and neck cancer cells stimulate tumor cell invasion through DNA-PK and Rac1 signaling. Clin. Cancer Res. 2016, 22, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Mallen-St Clair, J.; Alani, M.; Wang, M.B.; Srivatsan, E.S. Human papillomavirus in oropharyngeal cancer: The changing face of a disease. Biochim. Biophys. Acta 2016, 1866, 141–150. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spriggs, C.C.; Laimins, L.A. Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication. Viruses 2017, 9, 232. https://doi.org/10.3390/v9080232
Spriggs CC, Laimins LA. Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication. Viruses. 2017; 9(8):232. https://doi.org/10.3390/v9080232
Chicago/Turabian StyleSpriggs, Chelsey C., and Laimonis A. Laimins. 2017. "Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication" Viruses 9, no. 8: 232. https://doi.org/10.3390/v9080232
APA StyleSpriggs, C. C., & Laimins, L. A. (2017). Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication. Viruses, 9(8), 232. https://doi.org/10.3390/v9080232