
Investigations of Pro- and Anti-Apoptotic Factors Affecting African Swine Fever Virus Replication and Pathogenesis


Abstract
:
{kind=link}
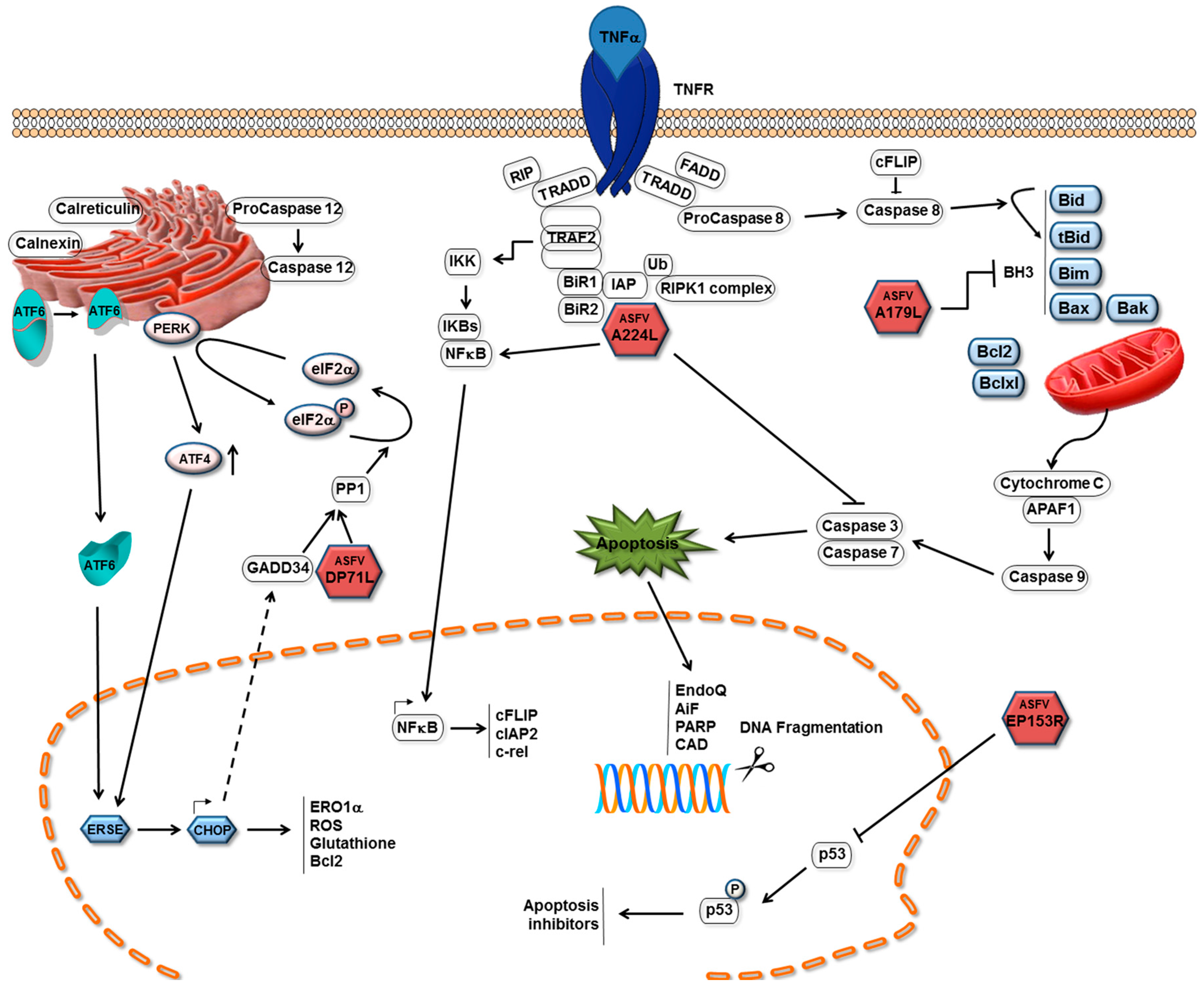{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction to African Swine Fever Virus
2. Induction of Apoptosis in Infected Cells
3. Inhibiting Apoptosis in the Infected Cell
3.1. Inhibitors of Apoptosis in ASFV-Infected Cells
The ASFV Bcl-2 Family Member A179L
3.2. The ASFV IAP-Family Member
3.2.1. Roles of Cellular IAPs
3.2.2. The ASFV IAP Protein A224L
3.3. ASFV Inhibition of the Stress-Activated Apoptosis Pathway
3.4. The ASFV C-Type Lectin Domain Containing Protein
4. Role of Apoptosis in Pathology and Immune Responses
Apoptosis in Tissue Samples from ASFV Infected Pigs
5. Conclusions and Future Work
Acknowledgments
Conflicts of Interest
References
- Smietanka, K.; Wozniakowski, G.; Kozak, E.; Niemczuk, K.; Fraczyk, M.; Bocian, L.; Kowalczyk, A.; Pejsak, Z. African Swine Fever Epidemic, Poland, 2014–2015. Emerg. Infect. Dis. 2016, 22, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Wozniakowski, G.; Kozak, E.; Kowalczyk, A.; Lyjak, M.; Pomorska-Mol, M.; Niemczuk, K.; Pejsak, Z. Current status of African swine fever virus in a population of wild boar in eastern Poland (2014–2015). Arch. Virol. 2016, 161, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, I.; Munoz, M.J.; Montes, F.; Perez, A.; Gogin, A.; Kolbasov, D.; de la Torre, A. Reproductive Ratio for the Local Spread of African Swine Fever in Wild Boars in the Russian Federation. Transbound. Emerg. Dis. 2016, 63, E237–E245. [Google Scholar] [CrossRef] [PubMed]
- OIE. World Animal Health Information Database (WAHID) 2017, World 731 Organisation for Animal Health (OIE). Available online: http://www.oie.int/wahis_2/public/wahid.php/Diseaseinformation/diseasehome (accessed on 2 August 2017).
- Andreani, J.; Bou Khalil, J.Y.; Sevvana, M.; Benamar, S.; Di Pinto, F.; Bitam, I.; Colson, P.; Klose, T.; Rossmann, M.G.; Raoult, D.; et al. Pacmanvirus, a new giant icosahedral virus at the crossroads between Asfarviridae and Faustoviruses. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bajrai, L.H.; Benamar, S.; Azhar, E.I.; Robert, C.; Levasseur, A.; Raoult, D.; la Scola, B. Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae. Viruses 2016, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Reteno, D.G.; Benamar, S.; Khalil, J.B.; Andreani, J.; Armstrong, N.; Klose, T.; Rossmann, M.; Colson, P.; Raoult, D.; La Scola, B. Faustovirus, an Asfarvirus-Related New Lineage of Giant Viruses Infecting Amoebae. J. Virol. 2015, 89, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- RamiroIbanez, F.; Ortega, A.; RuizGonzalvo, F.; Escribano, J.M.; Alonso, C. Modulation of immune cell populations and activation markers in the pathogenesis of African swine fever virus infection. Virus Res. 1997, 47, 31–40. [Google Scholar] [CrossRef]
- Galindo, I.; Hernaez, B.; Munoz-Moreno, R.; Cuesta-Geijo, M.A.; Dalmau-Mena, I.; Alonso, C. The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 2012, 3, e341. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, A.L.; Bustos, M.J.; Nogal, M.L.; de Buitrago, G.G.; Revilla, Y. Apoptosis induced in an early step of African swine fever virus entry into Vero cells does not require virus replication. Virology 2002, 294, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P. Enter the kill zone: Initiation of death signaling during virus entry. Virology 2011, 411, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P. Viruses and the Diversity of Cell Death. In Annual Review of Virology; Enquist, L.W., Ed.; Annual Reviews: Palo Alto, CA, USA, 2016; Volume 3, pp. 533–553. [Google Scholar]
- Alonso, C.; Miskin, J.; Hernaez, B.; Fernandez-Zapatero, P.; Soto, L.; Canto, C.; Rodriguez-Crespo, I.; Dixon, L.; Escribano, J.M. African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light-chain dynein. J. Virol. 2001, 75, 9819–9827. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Diaz-Gil, G.; Garcia-Gallo, M.; Quetglas, J.I.; Rodriguez-Crespo, I.; Dixon, L.; Escribano, J.M.; Alonso, C. The African swine fever virus dynein-binding protein p54 induces infected cell apoptosis. FEBS Lett. 2004, 569, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, B.; Escribano, J.M.; Alonso, C. Visualization of the African swine fever virus infection in living cells by incorporation into the virus particle of green fluorescent protein-p54 membrane protein chimera. Virology 2006, 350, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vallee, I.; Tait, S.W.G.; Powell, P.P. African swine fever virus infection of porcine aortic endothelial cells leads to inhibition of inflammatory responses, activation of the thrombotic state, and apoptosis. J. Virol. 2001, 75, 10372–10382. [Google Scholar] [CrossRef] [PubMed]
- Neilan, J.G.; Lu, Z.; Afonso, C.L.; Kutish, G.F.; Sussman, M.D.; Rock, D.L. An African Swine Fever Virus Gene with Similarity to the Protooncogene Bcl-2 and the Epstein-Barr-Virus Gene BHRF1. J. Virol. 1993, 67, 4391–4394. [Google Scholar] [PubMed]
- Neilan, J.G.; Lu, Z.; Kutish, G.F.; Zsak, L.; Burrage, T.G.; Borca, M.V.; Carrillo, C.; Rock, D.L. A BIR motif containing gene of African swine fever virus, 4CL, is nonessential for growth in vitro and viral virulence. Virology 1997, 230, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Yanez, R.J.; Rodriguez, J.M.; Nogal, M.L.; Yuste, L.; Enriquez, C.; Rodriguez, J.F.; Vinuela, E. Analysis of the Complete Nucleotide-Sequence of African Swine Fever Virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef] [PubMed]
- Bouillet, P.; Strasser, A. Bax and Bak: Back-bone of T cell death. Nat. Immunol. 2002, 3, 893–894. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. The structural biology of BH3-only proteins. Methods Enzymol. 2014, 544, 49–74. [Google Scholar] [PubMed]
- Afonso, C.L.; Neilan, J.G.; Kutish, G.F.; Rock, D.L. An African swine fever virus Bcl-2 homolog, 5-HL, suppresses apoptotic cell death. J. Virol. 1996, 70, 4858–4863. [Google Scholar] [PubMed]
- Hernaez, B.; Cabezas, M.; Munoz-Moreno, R.; Galindo, I.; Cuesta-Geijo, M.A.; Alonso, C. A179L, a New Viral Bcl2 Homolog Targeting Beclin 1 Autophagy Related Protein. Curr. Mol. Med. 2013, 13, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.; Rivas, C.; Esteban, M.; Escribano, J.M.; Alonso, C. African swine fever virus gene A179L, a viral homologue of Bcl-2, protects cells from programmed cell death. Virology 1996, 225, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Revilla, Y.; Cebrian, A.; Baixeras, E.; Martinez, C.; Vinuela, E.; Salas, M.L. Inhibition of apoptosis by the African swine fever virus Bcl-2 homologue: Role of the BH1 domain. Virology 1997, 228, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.; Rodriguez, F.; Escribano, J.M.; Alonso, C. Functionality and cell anchorage dependence of the African swine fever virus gene A179L, a viral Bcl-2 homolog, in insect cells. J. Virol. 1998, 72, 10227–10233. [Google Scholar] [PubMed]
- Galindo, I.; Hernaez, B.; Diaz-Gil, G.; Escribano, J.M.; Alonso, C. A 179L, a viral Bcl-2 homologue, targets the core Bcl-2 apoptotic machinery and its upstream BH3 activators with selective binding restrictions for Bid and Noxa. Virology 2008, 375, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Banjara, S.; Caria, S.; Dixon, L.K.; Hinds, M.G.; Kvansakul, M. Structural Insight into African Swine Fever Virus A179L-Mediated Inhibition of Apoptosis. J. Virol. 2017, 91, e02228-16. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J. Viral IAPs, then and now. Semin. Cell Dev. Biol. 2015, 39, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Byers, N.M.; Vandergaast, R.L.; Friesen, P.D. Baculovirus Inhibitor-of-Apoptosis Op-IAP3 Blocks Apoptosis by Interaction with and Stabilization of a Host Insect Cellular IAP. J. Virol. 2016, 90, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Vince, J. IAPs and Cell Death. In Apoptotic and Non-Apoptotic Cell Death; Nagata, S., Nakano, H., Eds.; Springer International Publishing AG: Cham, Switzerland, 2017; Volume 403, pp. 95–117. [Google Scholar]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.A.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Wayson, S.M.; Dixit, V.M.; Fairbrother, W.J.; Vucic, D. The inhibitor of apoptosis protein fusion c-IAP2 center dot MALT1 stimulates NF-κB activation independently of TRAF1 and TRAF2. J. Biol. Chem. 2006, 281, 29022–29029. [Google Scholar] [CrossRef] [PubMed]
- Mace, P.D.; Smits, C.; Vaux, D.L.; Silke, J.; Day, C.L. Asymmetric Recruitment of cIAPs by TRAF2. J. Mol. Biol. 2010, 400, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Kabaleeswaran, V.; Wang, Y.; Cheng, G.; Wu, H. Crystal Structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 Complexes: Affinity, Specificity, and Regulation. Mol. Cell 2010, 38, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Leo, E.; Stennicke, H.R.; Welsh, K.; Salvesen, G.S.; Reed, J.C. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999, 18, 5242–5251. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Stennicke, H.R.; Salvesen, G.S.; Reed, J.C. Endogenous inhibitors of caspases. J. Clin. Immunol. 1999, 19, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Deveraux, Q.; Tamm, I.; Welsh, K.; Assa-Munt, N.; Salvesen, G.S.; Reed, J.C. A single BIR domain of XIAP sufficient for inhibiting caspases. J. Biol. Chem. 1998, 273, 7787–7790. [Google Scholar] [CrossRef] [PubMed]
- Nogal, M.L.; de Buitrago, G.G.; Rodriguez, C.; Cubelos, B.; Carrascosa, A.L.; Salas, M.L.; Revilla, Y. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J. Virol. 2001, 75, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Chacon, M.R.; Almazan, F.; Nogal, M.L.; Vinuela, E.; Rodriguez, J.F. The African swine fever virus IAP homolog is a late structural polypeptide. Virology 1995, 214, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.I.; Nogal, M.L.; Carrascosa, A.L.; Salas, M.L.; Fresno, M.; Revilla, Y. African swine fever virus IAP-like protein induces the activation of nuclear factor κB. J. Virol. 2002, 76, 3936–3942. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.H.; Zeng, H.Q.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.Q.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2 α. J. Cell Biol. 2001, 153, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Barber, C.; Netherton, C.; Goatley, L.; Moon, A.; Goodbourn, S.; Dixon, L. Identification of residues within the African swine fever virus DP71L protein required for dephosphorylation of translation initiation factor eIF2 α and inhibiting activation of pro-apoptotic CHOP. Virology 2017, 504, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Abrams, C.; Hernaez, B.; Alcazar, A.; Escribano, J.M.; Dixon, L.; Alonso, C. The MyD116 African swine fever virus homologue interacts with the catalytic subunit of protein phosphatase 1 and activates its phosphatase activity. J. Virol. 2007, 81, 2923–2929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African Swine Fever Virus DP71L Protein Recruits the Protein Phosphatase 1 Catalytic Subunit To Dephosphorylate eIF2 α and Inhibits CHOP Induction but Is Dispensable for These Activities during Virus Infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, D.R.; Longnecker, R. The Herpes Simplex Virus Neurovirulence Factor gamma 34.5: Revealing Virus-Host Interactions. PLoS Pathog. 2016, 12, e1005449. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Oyadomari, S.; Novoa, I.; Lu, P.; Zhang, Y.H.; Harding, H.P.; Ron, D. Inhibition of a constitutive translation initiation factor 2 α phosphatase CReP, promotes survival of stressed cells. J. Cell Biol. 2003, 163, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Zsak, L.; Lu, Z.; Kutish, G.F.; Neilan, J.G.; Rock, D.L. An African swine fever virus virulence-associated gene NL-S with similarity to the herpes simplex virus ICP34.5 gene. J. Virol. 1996, 70, 8865–8871. [Google Scholar] [PubMed]
- Afonso, C.L.; Zsak, L.; Carrillo, C.; Borca, M.V.; Rock, D.L. African swine fever virus NL gene is not required for virus virulence. J. Gen. Virol. 1998, 79, 2543–2547. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Vasconcelos, G.; Dever, T.E. An eIF2 α-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2 α. Proc. Natl. Acad. Sci. USA 2015, 112, E3466–E3475. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; et al. ICP34.5 Protein of Herpes Simplex Virus Facilitates the Initiation of Protein Translation by Bridging Eukaryotic Initiation Factor 2 α (eIF2 α) and Protein Phosphatase 1. J. Biol. Chem. 2011, 286, 24785–24792. [Google Scholar] [CrossRef] [PubMed]
- Granja, A.G.; Nogal, M.L.; Hurtado, C.; Salas, J.; Salas, M.L.; Carrascosa, A.L.; Revilla, Y. Modulation of p53 cellular function and cell death by African swine fever virus. J. Virol. 2004, 78, 7165–7174. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, C.; Granja, A.G.; Bustos, M.J.; Nogal, M.L.; de Buitrago, G.G.; de Yebenes, V.G.; Salas, M.L.; Revilla, Y.; Carrascosa, A.L. The C-type lectin homologue gene (EP153R) of African swine fever virus inhibits apoptosis both in virus infection and in heterologous expression. Virology 2004, 326, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Blome, S.; Gabriel, C.; Beer, M. Pathogenesis of African swine fever in domestic pigs and European wild boar. Virus Res. 2013, 173, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Villamandos, J.C.; Bautista, M.J.; Sanchez-Cordon, P.J.; Carrasco, L. Pathology of African swine fever: The role of monocyte-macrophage. Virus Res. 2013, 173, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Gomezvillamandos, J.C.; Hervas, J.; Mendez, A.; Carrasco, L.; Delasmulas, J.M.; Villeda, C.J.; Wilkinson, P.J.; Sierra, M.A. Experimental African Swine Fever—Apoptosis of Lymphocytes and Virus-Replication in Other Cells. J. Gen. Virol. 1995, 76, 2399–2405. [Google Scholar] [CrossRef] [PubMed]
- RamiroIbanez, F.; Ortega, A.; Brun, A.; Escribano, J.M.; Alonso, C. Apoptosis: A mechanism of cell killing and lymphoid organ impairment during acute African swine fever virus infection. J. Gen. Virol. 1996, 77, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Zsak, L.; Neilan, J.G. Regulation of apoptosis in African swine fever virus-infected macrophages. Sci. World J. 2002, 2, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.; deLara, F.C.M.; delasMulas, J.M.; GomezVillamandos, J.C.; Perez, J.; Wilkinson, P.J.; Sierra, M.A. Apoptosis in lymph nodes in acute African swine fever. J. Comp. Pathol. 1996, 115, 415–428. [Google Scholar] [CrossRef]
- GomezVillamandos, J.C.; Hervas, J.; Moreno, C.; Carrasco, L.; Bautista, M.J.; Caballero, J.M.; Wilkinson, P.J.; Sierra, M.A. Subcellular changes in the tonsils of pigs infected with acute African swine fever virus. Vet. Res. 1997, 28, 179–189. [Google Scholar]
- Oura, C.A.L.; Powell, P.P.; Anderson, E.; Parkhouse, R.M.E. The pathogenesis of African swine fever in the resistant bushpig. J. Gen. Virol. 1998, 79, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Gomez Del Moral, M.; Ortuno, E.; Fernandez-Zapatero, P.; Alonso, F.; Alonso, C.; Ezquerra, A.; Dominguez, J. African swine fever virus infection induces tumor necrosis factor α production: Implications in pathogenesis. J. Virol. 1999, 73, 2173–2180. [Google Scholar] [PubMed]
- Salguero, F.J.; Ruiz-Villamor, E.; Bautista, M.J.; Sanchez-Cordon, P.J.; Carrasco, L.; Gomez-Villamandos, J.C. Changes in macrophages in spleen and lymph nodes during acute African swine fever: Expression of cytokines. Vet. Immunol. Immunopathol. 2002, 90, 11–22. [Google Scholar] [CrossRef]
- Salguero, F.J.; Sanchez-Cordon, P.J.; Sierra, M.A.; Jover, A.; Nunez, A.; Gomez-Villamandos, J.C. Apoptosis of thymocytes in experimental African swine fever virus infection. Histol. Histopathol. 2004, 19, 77–84. [Google Scholar] [PubMed]
- Fernandez de Marco, M.; Salguero, F.J.; Bautista, M.J.; Nunez, A.; Sanchez-Cordon, P.J.; Gomez-Villamandos, J.C. An immunohistochemical study of the tonsils in pigs with acute African swine fever virus infection. Res. Vet. Sci. 2007, 83, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Salguero, F.J.; Sanchez-Cordon, P.J.; Nunez, A.; de Marco, M.F.; Gomez-Villamandos, J.C. Proinflammatory cytokines induce lymphocyte apoptosis in acute African swine fever infection. J. Comp. Pathol. 2005, 132, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cordon, P.; Lorenzo Romero-Trevejo, J.; Pedrera, M.; Manuel Sanchez-Vizcaino, J.; Jose Bautista, M.; Carlos Gomez-Villamandos, J. Role of hepatic macrophages during the viral haemorrhagic fever induced by African Swine Fever Virus. Histol. Histopathol. 2008, 23, 683–691. [Google Scholar] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixon, L.K.; Sánchez-Cordón, P.J.; Galindo, I.; Alonso, C. Investigations of Pro- and Anti-Apoptotic Factors Affecting African Swine Fever Virus Replication and Pathogenesis. Viruses 2017, 9, 241. https://doi.org/10.3390/v9090241
Dixon LK, Sánchez-Cordón PJ, Galindo I, Alonso C. Investigations of Pro- and Anti-Apoptotic Factors Affecting African Swine Fever Virus Replication and Pathogenesis. Viruses. 2017; 9(9):241. https://doi.org/10.3390/v9090241
Chicago/Turabian StyleDixon, Linda K., Pedro J. Sánchez-Cordón, Inmaculada Galindo, and Covadonga Alonso. 2017. "Investigations of Pro- and Anti-Apoptotic Factors Affecting African Swine Fever Virus Replication and Pathogenesis" Viruses 9, no. 9: 241. https://doi.org/10.3390/v9090241
APA StyleDixon, L. K., Sánchez-Cordón, P. J., Galindo, I., & Alonso, C. (2017). Investigations of Pro- and Anti-Apoptotic Factors Affecting African Swine Fever Virus Replication and Pathogenesis. Viruses, 9(9), 241. https://doi.org/10.3390/v9090241