
Concomitant Presence of Hb Agrinio and - -Med Deletion in a Greek Male Patient with Hemoglobinopathy H: More Severe Phenotype and Literature Review
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
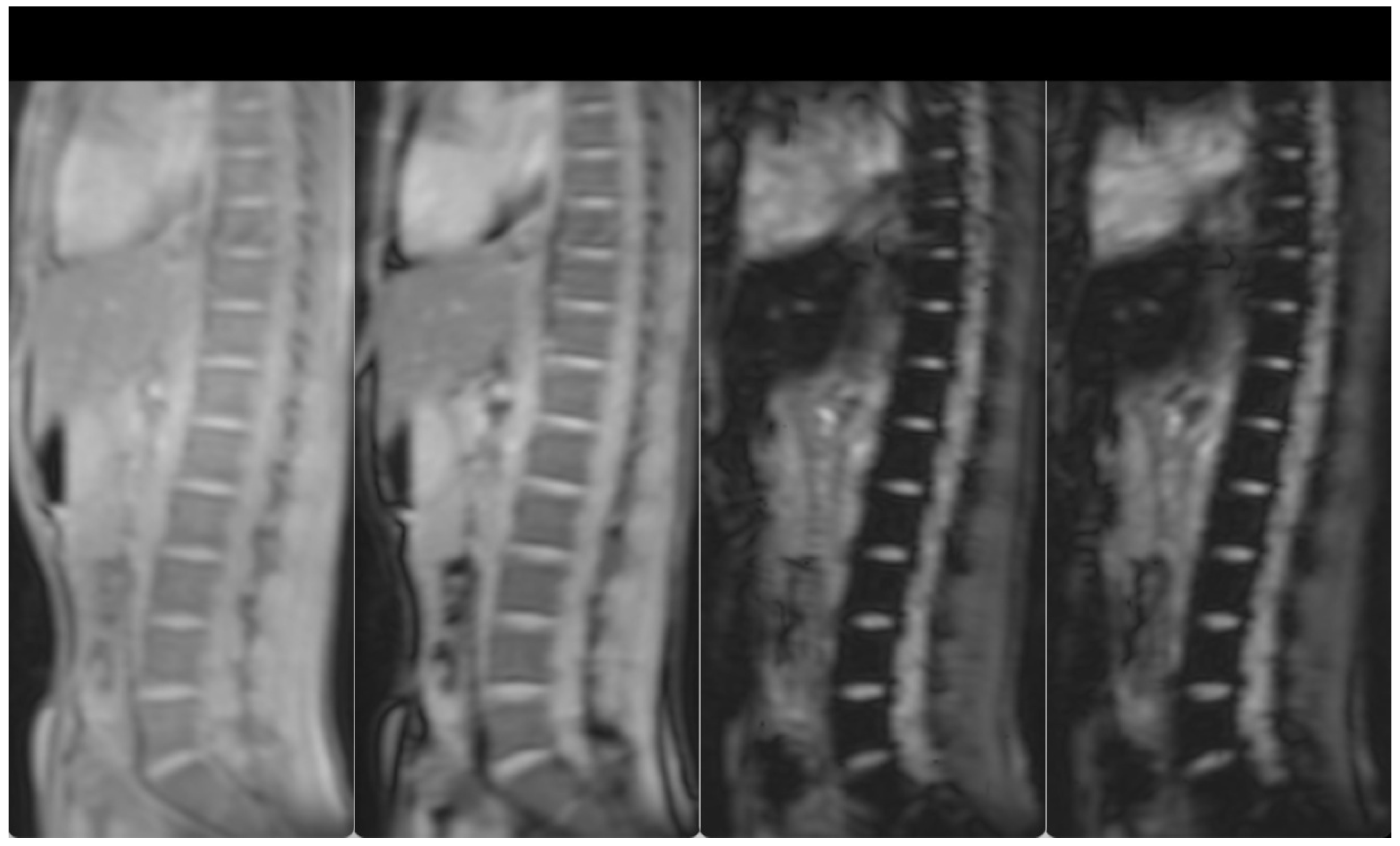
3. Case Presentation
4. Discussion and Literature Review
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galanello, R.; Cao, A. Gene test review. Alpha-thalassemia. Genet. Med. Off. J. Am. Coll. Med. Genet. 2011, 13, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Piel, F.B.; Weatherall, D.J. The α-thalassemias. N. Engl. J. Med. 2014, 371, 1908–1916. [Google Scholar] [CrossRef] [Green Version]
- Viprakasit, V.; Ekwattanakit, S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol./Oncol. Clin. N. Am. 2018, 32, 193–211. [Google Scholar] [CrossRef] [PubMed]
- Farashi, S.; Harteveld, C.L. Molecular basis of α-thalassemia. Blood Cells Mol. Dis. 2018, 70, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Traeger-Synodinos, J.; Metaxotou-Mavromati, A.; Kanavakis, E.; Vrettou, C.; Papassotiriou, I.; Michael, T.; Kattamis, C. An α-thalassemic hemoglobinopathy: Homozygosity for the HB Agrinio α 2-globin chain variant. Hemoglobin 1998, 22, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Traeger-Synodinos, J.; Douna, V.; Papassotiriou, I.; Stamoulakatou, A.; Ladis, V.; Siahanidou, T.; Fylaktou, I.; Kanavakis, E. Variable and often severe phenotypic expression in patients with the α-thalassemic variant Hb Agrinio [α29(B10)Leu→Pro (α2)]. Hemoglobin 2010, 34, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Felekis, X.; Phylactides, M.; Drousiotou, A.; Christou, S.; Kyrri, A.; Kyriakou, K.; Kalogerou, E.; Christopoulos, G.; Kleanthous, M. Hb Agrinio [α29(B10)Le-->uPro (α2)] in combination with --MED I. Results in a severe form of Hb H disease. Hemoglobin 2008, 32, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Wajcman, H.; Traeger-Synodinos, J.; Papassotiriou, I.; Giordano, P.C.; Harteveld, C.L.; Baudin-Creuza, V.; Old, J. Unstable and thalassemic α chain hemoglobin variants: A cause of Hb H disease and thalassemia intermedia. Hemoglobin 2008, 32, 327–349. [Google Scholar] [CrossRef] [PubMed]
- Szepetowski, S.; Berger, C.; Joly, P.; Baron-Joly, S.; Huguenin, Y.; Cantais, A.; Brun, S.; Ged, C.; Badens, C.; Thuret, I.; et al. Homozygosity for the hyperunstable hemoglobin variant Hb Agrinio (HBA2:c.89T>C) leads to severe antenatal anemia: Eight new cases in three families. Am. J. Hematol. 2022, 97, E393–E395. [Google Scholar] [CrossRef] [PubMed]
- Kanavakis, E.; Papassotiriou, I.; Karagiorga, M.; Vrettou, C.; Metaxotou-Mavrommati, A.; Stamoulakatou, A.; Kattamis, C.; Traeger-Synodinos, J. Phenotypic and molecular diversity of haemoglobin H disease: A Greek experience. Br. J. Haematol. 2000, 111, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Douna, V.; Papassotiriou, I.; Stamoulakatou, A.; Metaxotou-Mavrommati, A.; Kanavakis, E.; Traeger-Synodinos, J. Association of mild and severely unstable α chain variants: The first observation of a compound heterozygote with Hb Setif [α94(G1)Asp-->Tyr (α2)] and Hb Agrinio [α29(B10)Leu-->Pro (α2)] in a Greek family. Hemoglobin 2008, 32, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Dimishkovska, M.; Kuzmanovska, M.; Kocheva, S.; Martinova, K.; Karanfilski, O.; Stojanoski, Z.; Plaseska-Karanfilska, D. First Cases of Hb Agrinio Described in Patients from the Republic of Macedonia. Hemoglobin 2017, 41, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, M.; Drakoulakou, O.; Papapanagiotou, E.; Pessini, D.; Loutradi-Anagnostou, A. Hb A2-Agrinio [δ43(CD2)Glu-->Gly(GAG-->GGG)]: A new δ chain variant detected in a Greek family. Hemoglobin 1995, 19, 295–299. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Gonzalo, F.; Baiget, M.; Badell, I.; Ricard, P.; Vinuesa, L.; Martínez-Nieto, J.; Ropero, P.; Villegas, A.; González, F.A.; Díaz-Mediavilla, J.; et al. Study of three families with Hb Agrinio [α29(B10)Leu→Pro, CTG>CCG (α2)] in the Spanish population: Three homozygous cases. Hemoglobin 2012, 36, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Kanavakis, E.; Traeger-Synodinos, J.; Papasotiriou, I.; Vrettou, C.; Metaxotou-Mavromati, A.; Stamoulakatou, A.; Lagona, E.; Kattamis, C. The interaction of α zero thalassaemia with Hb Icaria: Three unusual cases of haemoglobinopathy H. Br. J. Haematol. 1996, 92, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Tampaki, A.; Theodoridou, S.; Apostolou, C.; Delaki, E.E.; Vlachaki, E. A case of late diagnosis of compound heterozygosity for Hb Adana (HBA2:c.179G>A) in trans to an α+- thalassemia deletion: Guilty or innocent. Hippokratia 2020, 24, 43–45. [Google Scholar] [PubMed]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Cappellini, M.D.; Kattamis, A.; Voskaridou, E.; Perrotta, S.; Piga, A.G.; Filosa, A.; Porter, J.B.; Coates, T.D.; Forni, G.L.; et al. Luspatercept for the treatment of anaemia in non-transfusion-dependent β-thalassaemia (BEYOND): A phase 2, randomised, double-blind, multicentre, placebo-controlled trial. Lancet. Haematol. 2022, 9, e733–e744. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Hematological and Biochemical Index | |
|---|---|
| Hgb | 8.1 gr/dL |
| WBC | 4.01 k/μL |
| PLT | 152.0 k/μL |
| FERRITIN | 644.8 ng/mL |
| LDH | 1821 IU/lt |
| TBIL | 3.9 mg/dL |
| URIC ACID | 4.1 mg/dL |
| UREA | 52 mg/dL |
| CREATINE | 1.0 mg/dL |
| GLUCOSE | 111 mg/dL |
| Reference | Country | Number of Cases | Molecular Defects | Clinical Manifestations |
|---|---|---|---|---|
| (Galanello et al. 2011) [1] (de la Fuente-Gonzalo et al. 2012) [14] | Spain (Catalonia, Andalusia, Madrid) | 14 cases (from 3 families, with the 2 of them being of Gypsy ethnicity) | a2 with mutation at codon 29 (CTG > CCG) | -11 cases of heterozygous state-silent phenotype of thalassemia without anemia and mild microcytosis with iron deficiency -3 cases of homozygous state-severe intermediate phenotype HbH disease |
| (Dimishkovska et al. 2017) [12] | North Macedonia | 2 cases (from 2 unrelated families of Romani ethnicity) | a2 with mutation at codon 29 (CTG > CCG) [a29(B10) Leu to Pro; HBA2: c.89T > C; aAgrinioa/] | 2 cases of homozygous state-severe or intermediate phenotype for HbH disease with blood transfusions since infancy and hemolytic anemia |
| (Felekis et al. 2008) [7] | Cyprus | 2 cases (from 2 Greek-Cypriot families) | - -Med and Hb Agrinio [a29(B10)Leu to Pro (a2)] | 2 cases with compound heterozygosity-severe or intermediateHbH disease with need for regular transfusions from early age |
| (Douna et al. 2008) [11] | Greece | 1 case | Hb Setif, a mutation of a2-globin at codon 94, [α94(G1)Asp→Tyr, GAC > TAC (α2)], and Hb Agrinio, mutation a2-globin at codon 29, [a29(B10) Leu to Pro; CTG > CCG (a2)] | 1 case with compound heterozygosity-expression of mild anemia, with no need for regular transfusions and normal development |
| (Szepetowski et al. 2022) [9] | -Spain (2 different families with gypsy ethnicity) -Bulgaria (1 family) | 8 cases | Homozygous Hb Agrinio [a29(B10) Leu to Pro; HBA2: c.89T > C; aAgrinioa] | Severe or intermediate phenotype for HbH disease with blood transfusions since infancy and hemolytic anemia or even hydrops fetalis |
| (Traeger-Synodinos et al. 2010) [6] | Greece | -12 cases homozygous/compound heterozygous -over 25 cases of single heterozygocity (from a 15 year survey) | -Hb Agrinio mutation (αAgrinioα/αAgrinioα) -Hb Agrinio and the polyadenylation signal (polyA)site mutation (AATAAA > AATAAG, HBA2:c.*+94A > G or αPAα -Hb Agrinio and the α0 deletion Med -Hb Agrinio and the α2-globin gene IVS-I donor site pentanucleotide Hemoglobin deletion (HBA2:c.95 + 2_95 + 6delTGAGG or αHphα) -Hb Agrinio and Hb Setif, [α94(G1)Asp→Tyr, GAC > TAC(α2) or HBA2:c.283G >T]. | -4 cases of homozygous state-severe or intermediate phenotype for HbH disease with blood transfusions from early infancy and hemolytic anemia or even hydrops fetalis -8 cases of compound heterozygosity-wide range of phenotypic severity from mild thalassemia with no transfusions and mild anemia to regularly blood transfusions every 15 days, splenectomy and development deficiency |
| (Kanavakis et al. 1996) [15] | Greece | 3 cases | - -Med/aICariaa | 3 cases of compound heterozygosity for Hb Icaria-clinically expressing a severe HbH disease with regular need for blood transfusions from an early age, development deficiency and splenomegaly |
| (Tampaki et al. 2020) [16] | Greece | 1 case | Hb Adana (aadanaa) (HBA2:c.179G>A or HBA1) (-a3.7 kb deletion/Hb Adana) | Chronic hemolytic anemia and significant splenomegaly, thrombocytopenia with no blood transfusions |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diamantidis, M.D.; Pitsava, S.; Zayed, O.; Argyrakouli, I.; Karapiperis, K.; Chatzoulis, C.; Alexiou, E.; Manafas, A.; Tsangalas, E.; Karakoussis, K. Concomitant Presence of Hb Agrinio and - -Med Deletion in a Greek Male Patient with Hemoglobinopathy H: More Severe Phenotype and Literature Review. Hematol. Rep. 2023, 15, 483-490. https://doi.org/10.3390/hematolrep15030050
Diamantidis MD, Pitsava S, Zayed O, Argyrakouli I, Karapiperis K, Chatzoulis C, Alexiou E, Manafas A, Tsangalas E, Karakoussis K. Concomitant Presence of Hb Agrinio and - -Med Deletion in a Greek Male Patient with Hemoglobinopathy H: More Severe Phenotype and Literature Review. Hematology Reports. 2023; 15(3):483-490. https://doi.org/10.3390/hematolrep15030050
Chicago/Turabian StyleDiamantidis, Michael D., Stefania Pitsava, Omar Zayed, Ioanna Argyrakouli, Konstantinos Karapiperis, Christos Chatzoulis, Evangelos Alexiou, Achilles Manafas, Evangelos Tsangalas, and Konstantinos Karakoussis. 2023. "Concomitant Presence of Hb Agrinio and - -Med Deletion in a Greek Male Patient with Hemoglobinopathy H: More Severe Phenotype and Literature Review" Hematology Reports 15, no. 3: 483-490. https://doi.org/10.3390/hematolrep15030050