
Treatment of Immune Thrombocytopenia: Contextualization from a Historical Perspective

 , ,
, ,
Abstract
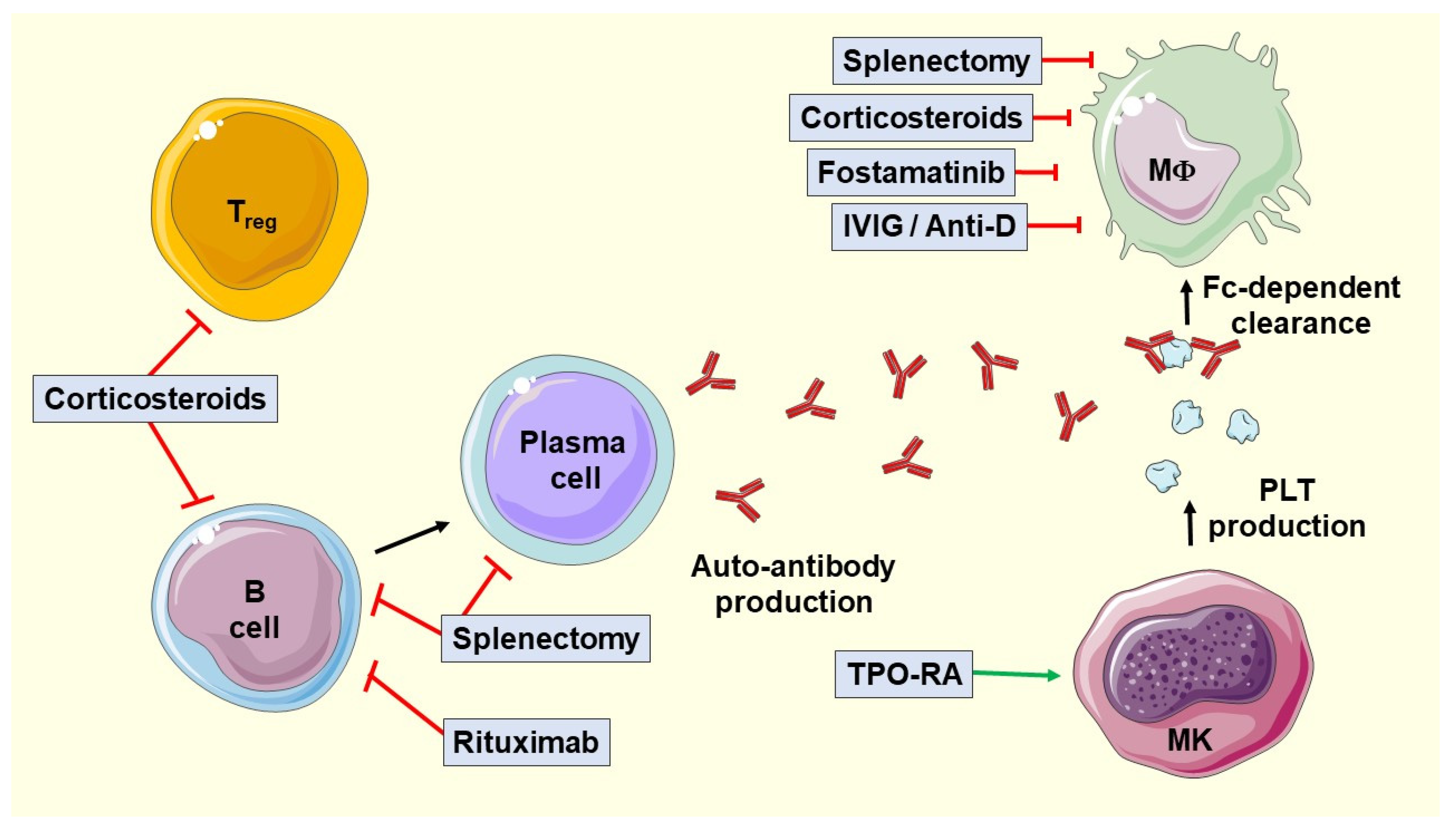
:1. Introduction
2. Werlhof’s Disease
3. Current Treatment Modalities
4. Treatment Indications
5. First-Line Treatment
5.1. Glucocorticoids
5.2. Intravenous Immunoglobulins (IVIGs)
5.3. Anti-D Immunoglobulin
6. Second-Line Treatment
6.1. Thrombopoietin Receptor Agonists
6.1.1. Adverse Events
6.1.2. Agonist Change (Switch)
6.1.3. TPO-RA Discontinuation
6.1.4. Avatrombopag
6.2. Fostamatinib
6.3. Rituximab
6.4. Splenectomy
7. Refractory ITP
8. New Therapies for ITP
8.1. Fine Tuning the Immune Response
8.2. Fine Tuning the Immune Response and Promoting Megakaryopoiesis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rodeghiero, F.; Stasi, R.; Gernsheimer, T.; Michel, M.; Provan, D.; Arnold, D.M.; Bussel, J.B.; Cines, D.B.; Chong, B.H.; Cooper, N.; et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: Report from an international working group. Blood 2009, 113, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Terrell, D.R.; Beebe, L.A.; Vesely, S.K.; Neas, B.R.; Segal, J.B.; George, J.N. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am. J. Hematol. 2010, 85, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, P.E.; Hall, S.A.; Feudjo-Tepie, M.; Mitrani-Gold, F.S.; Logie, J. The incidence of idiopathic thrombocytopenic purpura among adults: A population-based study and literature review. Eur. J. Haematol. 2009, 83, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Hedman, A.; Henter, J.I.; Hedlund, I.; Elinder, G. Prevalence and treatment of chronic idiopathic thrombocytopenic purpura of childhood in Sweden. Acta Paediatr. 1997, 86, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Uzun, G.; Bakchoul, T. Primary Immune Thrombocytopenia: Novel Insights into Pathophysiology and Disease Management. J. Clin. Med. 2021, 10, 789. [Google Scholar] [CrossRef] [PubMed]
- Consolini, R.; Legitimo, A.; Caparello, M.C. The Centenary of Immune Thrombocytopenia—Part 1: Revising Nomenclature and Pathogenesis. Front. Pediatr. 2016, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- González-López, T.J.; Newland, A.; Provan, D. Current Concepts in the Diagnosis and Management of Adult Primary Immune Thrombocytopenia: Our Personal View. Medicina 2023, 59, 815. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carballeira, D.; Bernardo, Á.; Caro, A.; Soto, I.; Gutiérrez, L. Pathophysiology, Clinical Manifestations and Diagnosis of Immune Thrombocytopenia: Contextualization from a Historical Perspective. Hematol. Rep. 2024, 16, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Newland, A.C. ITP: A historical perspective. Br. J. Haematol. 2011, 153, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Imbach, P.; Kühne, T.; Signer, E. Historical aspects and present knowledge of idiopathic thrombocytopenic purpura. Br. J. Haematol. 2002, 119, 894–900. [Google Scholar] [CrossRef]
- Mingot-Castellano, M.E.; Canaro Hirnyk, M.; Sánchez-González, B.; Álvarez-Román, M.T.; Bárez-García, A.; Bernardo-Gutiérrez, Á.; Bernat-Pablo, S.; Bolaños-Calderón, E.; Butta-Coll, N.; Caballero-Navarro, G.; et al. Recommendations for the Clinical Approach to Immune Thrombocytopenia: Spanish ITP Working Group (GEPTI). J. Clin. Med 2023, 12, 6422. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Terrell, D.R.; Arnold, D.M.; Buchanan, G.; Cines, D.B.; Cooper, N.; Cuker, A.; Despotovic, J.M.; George, J.N.; Grace, R.F.; et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019, 3, 3829–3866. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Arnold, D.M.; Bussel, J.B.; Chong, B.H.; Cooper, N.; Gernsheimer, T.; Ghanima, W.; Godeau, B.; González-López, T.J.; Grainger, J.; et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019, 3, 3780–3817. [Google Scholar] [CrossRef] [PubMed]
- Neunert, C.; Noroozi, N.; Norman, G.; Buchanan, G.R.; Goy, J.; Nazi, I.; Kelton, J.G.; Arnold, D.M. Severe bleeding events in adults and children with primary immune thrombocytopenia: A systematic review. J. Thromb. Haemost. 2015, 13, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.C.; Djulbegovic, B.; Shamai-Lubovitz, O.; Mozes, B. The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. Arch. Intern. Med. 2000, 160, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Adelborg, K.; Kristensen, N.R.; Nørgaard, M.; Bahmanyar, S.; Ghanima, W.; Kilpatrick, K.; Frederiksen, H.; Ekstrand, C.; Sørensen, H.T.; Fynbo Christiansen, C. Cardiovascular and bleeding outcomes in a population-based cohort of patients with chronic immune thrombocytopenia. J. Thromb. Haemost. 2019, 17, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Piel-Julian, M.L.; Mahévas, M.; Germain, J.; Languille, L.; Comont, T.; Lapeyre-Mestre, M.; Payrastre, B.; Beyne-Rauzy, O.; Michel, M.; Godeau, B.; et al. Risk factors for bleeding, including platelet count threshold, in newly diagnosed immune thrombocytopenia adults. J. Thromb. Haemost. 2018, 16, 1830–1842. [Google Scholar] [CrossRef] [PubMed]
- Wintrobe, M.M.; Cartwright, G.E.; Palmer, J.G.; Kuhns, W.J.; Samuels, L.T. Effect of corticotrophin and cortisone on the blood in various disorders in man. AMA Arch. Intern. Med. 1951, 88, 310–336. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.C. Response of Resistant Idiopathic Thrombocytopenic Purpura to Pulsed High-Dose Dexamethasone Therapy. N. Engl. J. Med. 1994, 330, 1560–1564. [Google Scholar] [CrossRef]
- Zufferey, A.; Kapur, R.; Semple, J. Pathogenesis and Therapeutic Mechanisms in Immune Thrombocytopenia (ITP). J. Clin. Med. 2017, 6, 16. [Google Scholar] [CrossRef]
- Mithoowani, S.; Gregory-Miller, K.; Goy, J.; Miller, M.C.; Wang, G.; Noroozi, N.; Kelton, J.G.; Arnold, D.M. High-dose dexamethasone compared with prednisone for previously untreated primary immune thrombocytopenia: A systematic review and meta-analysis. Lancet Haematol. 2016, 3, e489–e496. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Fu, L.; Chen, Z.; Gu, H.; Ma, J.; Wu, R. High-dose dexamethasone as a replacement for traditional prednisone as the first-line treatment in children with previously untreated primary immune thrombocytopenia: A prospective, randomized single-center study. Int. J. Hematol. 2020, 112, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.; Ghanima, W. Immune Thrombocytopenia. N. Engl. J. Med. 2019, 381, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, L.; Hao, H.; Jansen, A.J.G.; Liu, G.; Li, H.; Liu, X.; Zhao, Y.; Peng, J.; Hou, M. First line treatment of adult patients with primary immune thrombocytopenia: A real-world study. Platelets 2020, 31, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Imbach, P. Treatment of immune thrombocytopenia with intravenous immunoglobulin and insights for other diseases. Swiss. Med. Wkly. 2012, 142, w13593. [Google Scholar] [CrossRef]
- Imbach, P.; Barandun, S.; d’Apuzzo, V.; Baumgartner, C.; Hirt, A.; Morell, A.; Rossi, E.; Schöni, M.; Vest, M.; Wagner, H.P. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981, 1, 1228–1231. [Google Scholar] [CrossRef]
- Newland, A.C.; Treleaven, J.G.; Minchinton, R.M.; Waters, A.H. High-dose intravenous IgG in adults with autoimmune thrombocytopenia. Lancet 1983, 1, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Newland, A.C.; Boots, M.A.; Patterson, K.G. Intravenous IgG for autoimmune thrombocytopenia in pregnancy. N. Engl. J. Med. 1984, 310, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Crow, A.R.; Lazarus, A.H. The Mechanisms of Action of Intravenous Immunoglobulin and Polyclonal Anti-D Immunoglobulin in the Amelioration of Immune Thrombocytopenic Purpura: What Do We Really Know? Transfus. Med. Rev. 2008, 22, 103–116. [Google Scholar] [CrossRef]
- Provan, D.; Stasi, R.; Newland, A.C.; Blanchette, V.S.; Bolton-Maggs, P.; Bussel, J.B.; Chong, B.H.; Cines, D.B.; Gernsheimer, T.B.; Godeau, B.; et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010, 115, 168–186. [Google Scholar] [CrossRef]
- Godeau, B.; Chevret, S.; Varet, B.; Lefrère, F.; Zini, J.M.; Bassompierre, F.; Chèze, S.; Legouffe, E.; Hulin, C.; Grange, M.J.; et al. Intravenous immunoglobulin or high-dose methylprednisolone, with or without oral prednisone, for adults with untreated severe autoimmune thrombocytopenic purpura: A randomised, multicentre trial. Lancet 2002, 359, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Go, R.S.; Johnston, K.L.; Bruden, K.C. The association between platelet autoantibody specificity and response to intravenous immunoglobulin G in the treatment of patients with immune thrombocytopenia. Haematologica 2007, 92, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Ma, S.H.; Liu, J.; Hou, Y.; Liu, X.M.; Niu, T.; Xu, R.R.; Guo, C.S.; Wang, X.M.; Cheng, Y.F.; et al. Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: A multicenter cohort study. J. Thromb. Haemost. 2014, 12, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; van der Wal, D.E.; Zhu, G.; Xu, M.; Yougbare, I.; Ma, L.; Vadasz, B.; Carrim, N.; Grozovsky, R.; Ruan, M.; et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat. Commun. 2015, 6, 7737. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Rosovsky, R.P.; Karp Leaf, R.S.; Smith, D.B.; Goodarzi, K.; Fogerty, A.E.; Sykes, D.B.; Kuter, D.J. A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv. 2020, 4, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Kiefel, V.; Amberg, R.; Mueller-Eckhardt, C. Treatment of autoimmune thrombocytopenic purpura with rhesus antibodies (anti-Rh0(D). Blut 1984, 49, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.; Mueller-Eckhardt, C.; Kiefel, V. Effect of intravenous immunoglobulin in immune thrombocytopenia. Lancet 1983, 2, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Graziano, J.N.; Kimberly, R.P.; Pahwa, S.; Aledort, L.M. Intravenous anti-D treatment of immune thrombocytopenic purpura: Analysis of efficacy, toxicity, and mechanism of effect. Blood 1991, 77, 1884–1893. [Google Scholar] [CrossRef] [PubMed]
- Cooper, N.; Woloski, B.M.R.; Fodero, E.M.; Novoa, M.; Leber, M.; Beer, J.H.; Bussel, J.B. Does treatment with intermittent infusions of intravenous anti-D allow a proportion of adults with recently diagnosed immune thrombocytopenic purpura to avoid splenectomy? Blood 2002, 99, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Kane, I.; Ragucci, D.; Shatat, I.F.; Bussel, J.; Kalpatthi, R. Comparison of intravenous immune globulin and high dose anti-D immune globulin as initial therapy for childhood immune thrombocytopenic purpura. Br. J. Haematol. 2010, 149, 79–83. [Google Scholar] [CrossRef]
- Michel, M.; Novoa, M.V.; Bussel, J.B. Intravenous anti-D as a treatment for immune thrombocytopenic purpura (ITP) during pregnancy. Br. J. Haematol. 2003, 123, 142–146. [Google Scholar] [CrossRef]
- Newman, G.C.; Novoa, M.V.; Fodero, E.M.; Lesser, M.L.; Woloski, B.M.; Bussel, J.B. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br. J. Haematol. 2001, 112, 1076–1078. [Google Scholar] [CrossRef]
- Scaradavou, A.; Woo, B.; Woloski, B.M.; Cunningham-Rundles, S.; Ettinger, L.J.; Aledort, L.M.; Bussel, J.B. Intravenous anti-D treatment of immune thrombocytopenic purpura: Experience in 272 patients. Blood 1997, 89, 2689–2700. [Google Scholar] [CrossRef]
- Lazarus, A.H.; Crow, A.R. Mechanism of action of IVIG and anti-D in ITP. Transfus. Apher. Sci. 2003, 28, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Naithani, R.; Kumar, R.; Mahapatra, M.; Tyagi, S.; Saxena, R. Efficacy and safety of anti-D for treatment of adults with immune thrombocytopenia. Platelets 2009, 20, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.; Liebman, H.A. Anti-RhD immunoglobulin in the treatment of immune thrombocytopenia. Biologics 2009, 3, 57–62. [Google Scholar] [PubMed]
- Gaines, A.R. Disseminated intravascular coagulation associated with acute hemoglobinemia or hemoglobinuria following Rh(0)(D) immune globulin intravenous administration for immune thrombocytopenic purpura. Blood 2005, 106, 1532–1537. [Google Scholar] [CrossRef]
- Tarantino, M.D.; Bussel, J.B.; Cines, D.B.; McCrae, K.R.; Gernsheimer, T.; Liebman, H.A.; Wong, W.-Y.; Kulkarni, R.; Grabowski, E.; McMillan, R. A closer look at intravascular hemolysis (IVH) following intravenous anti-D for immune thrombocytopenic purpura (ITP). Blood 2007, 109, 5527. [Google Scholar] [CrossRef]
- Kaushansky, K. Thrombopoiesis. Semin. Hematol. 2015, 52, 4–11. [Google Scholar] [CrossRef]
- Bartley, T.D.; Bogenberger, J.; Hunt, P.; Li, Y.S.; Lu, H.S.; Martin, F.; Chang, M.S.; Samal, B.; Nichol, J.L.; Swift, S. Identification and cloning of a megakaryocyte growth and development factor that is a ligand for the cytokine receptor Mpl. Cell 1994, 77, 1117–1124. [Google Scholar] [CrossRef]
- Kuter, D.J.; Beeler, D.L.; Rosenberg, R.D. The purification of megapoietin: A physiological regulator of megakaryocyte growth and platelet production. Proc. Natl. Acad. Sci. USA 1994, 91, 11104–11108. [Google Scholar] [CrossRef]
- Lok, S.; Kaushansky, K.; Holly, R.D.; Kuijper, J.L.; Lofton-Day, C.E.; Oort, P.J.; Grant, F.J.; Heipel, M.D.; Burkhead, S.K.; Kramer, J.M. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature 1994, 369, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J. The biology of thrombopoietin and thrombopoietin receptor agonists. Int. J. Hematol. 2013, 98, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Emmons, R.V.; Reid, D.M.; Cohen, R.L.; Meng, G.; Young, N.S.; Dunbar, C.E.; Shulman, N.R. Human thrombopoietin levels are high when thrombocytopenia is due to megakaryocyte deficiency and low when due to increased platelet destruction. Blood 1996, 87, 4068–4071. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Kurata, Y.; Tomiyama, Y.; Tahara, T.; Kato, T.; Tadokoro, S.; Shiraga, M.; Honda, S.; Kanakura, Y.; Matsuzawa, Y. Circulating thrombopoietin level in chronic immune thrombocytopenic purpura. Br. J. Haematol. 1996, 93, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W.; Cooper, N.; Rodeghiero, F.; Godeau, B.; Bussel, J.B. Thrombopoietin receptor agonists: Ten years later. Haematologica 2019, 104, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Nplate Epar. Available online: https://www.ema.europa.eu/en/documents/product-information/nplate-epar-product-information_en.pdf (accessed on 11 February 2024).
- Revolade Epar. Available online: https://www.ema.europa.eu/en/documents/product-information/revolade-epar-product-information_en.pdf (accessed on 11 February 2024).
- Rodeghiero, F.; Carli, G. Beyond immune thrombocytopenia: The evolving role of thrombopoietin receptor agonists. Ann. Hematol. 2017, 96, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Bussel, J.B.; Lyons, R.M.; Pullarkat, V.; Gernsheimer, T.B.; Senecal, F.M.; Aledort, L.M.; George, J.N.; Kessler, C.M.; Sanz, M.A.; et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: A double-blind randomised controlled trial. Lancet 2008, 371, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Bussel, J.B.; Newland, A.; Baker, R.I.; Lyons, R.M.; Wasser, J.; Viallard, J.-F.; Macik, G.; Rummel, M.; Nie, K.; et al. Long-term treatment with romiplostim in patients with chronic immune thrombocytopenia: Safety and efficacy. Br. J. Haematol. 2013, 161, 411–423. [Google Scholar] [CrossRef]
- Cines, D.B.; Wasser, J.; Rodeghiero, F.; Chong, B.H.; Steurer, M.; Provan, D.; Lyons, R.; Garcia-Chavez, J.; Carpenter, N.; Wang, X.; et al. Safety and efficacy of romiplostim in splenectomized and nonsplenectomized patients with primary immune thrombocytopenia. Haematologica 2017, 102, 1342–1351. [Google Scholar] [CrossRef]
- Bussel, J.B.; Provan, D.; Shamsi, T.; Cheng, G.; Psaila, B.; Kovaleva, L.; Salama, A.; Jenkins, J.M.; Roychowdhury, D.; Mayer, B.; et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 641–648. [Google Scholar] [CrossRef]
- Cheng, G.; Saleh, M.N.; Marcher, C.; Vasey, S.; Mayer, B.; Aivado, M.; Arning, M.; Stone, N.L.; Bussel, J.B. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): A 6-month, randomised, phase 3 study. Lancet 2011, 377, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.M.; Saleh, M.N.; Khelif, A.; Salama, A.; Portella, M.S.O.; Burgess, P.; Bussel, J.B. Safety and efficacy of long-term treatment of chronic/persistent ITP with eltrombopag: Final results of the EXTEND study. Blood 2017, 130, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; Buchanan, G.R.; Nugent, D.J.; Gnarra, D.J.; Bomgaars, L.R.; Blanchette, V.S.; Wang, Y.-M.; Nie, K.; Jun, S. A randomized, double-blind study of romiplostim to determine its safety and efficacy in children with immune thrombocytopenia. Blood 2011, 118, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Elalfy, M.S.; Abdelmaksoud, A.A.; Eltonbary, K.Y. Romiplostim in children with chronic refractory ITP: Randomized placebo controlled study. Ann. Hematol. 2011, 90, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, M.D.; Bussel, J.B.; Blanchette, V.S.; Beam, D.; Roy, J.; Despotovic, J.; Raj, A.; Carpenter, N.; Mehta, B.; Eisen, M. Long-term treatment with romiplostim and treatment-free platelet responses in children with chronic immune thrombocytopenia. Haematologica 2019, 104, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, M.D.; Bussel, J.B.; Blanchette, V.S.; Despotovic, J.; Bennett, C.; Raj, A.; Williams, B.; Beam, D.; Morales, J.; Rose, M.J.; et al. Romiplostim in children with immune thrombocytopenia: A phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016, 388, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Bussel, J.B.; de Miguel, P.G.; Despotovic, J.M.; Grainger, J.D.; Sevilla, J.; Blanchette, V.S.; Krishnamurti, L.; Connor, P.; David, M.; Boayue, K.B.; et al. Eltrombopag for the treatment of children with persistent and chronic immune thrombocytopenia (PETIT): A randomised, multicentre, placebo-controlled study. Lancet Haematol. 2015, 2, e315–e325. [Google Scholar] [CrossRef] [PubMed]
- Grainger, J.D.; Locatelli, F.; Chotsampancharoen, T.; Donyush, E.; Pongtanakul, B.; Komvilaisak, P.; Sosothikul, D.; Drelichman, G.; Sirachainan, N.; Holzhauer, S.; et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): A randomised, multicentre, placebo-controlled trial. Lancet 2015, 386, 1649–1658. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Stasi, R.; Giagounidis, A.; Viallard, J.-F.; Godeau, B.; Pabinger, I.; Cines, D.; Liebman, H.; Wang, X.; Woodard, P. Long-term safety and tolerability of romiplostim in patients with primary immune thrombocytopenia: A pooled analysis of 13 clinical trials. Eur. J. Haematol. 2013, 91, 423–436. [Google Scholar] [CrossRef]
- González-Porras, J.R.; Mingot-Castellano, M.E.; Andrade, M.M.; Alonso, R.; Caparrós, I.; Arratibel, M.C.; Fernández-Fuertes, F.; Cortti, M.J.; Pascual, C.; Sánchez-González, B.; et al. Use of eltrombopag after romiplostim in primary immune thrombocytopenia. Br. J. Haematol. 2015, 169, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Khellaf, M.; Viallard, J.-F.; Hamidou, M.; Cheze, S.; Roudot-Thoraval, F.; Lefrere, F.; Fain, O.; Audia, S.; Abgrall, J.-F.; Michot, J.-M.; et al. A retrospective pilot evaluation of switching thrombopoietic receptor-agonists in immune thrombocytopenia. Haematologica 2013, 98, 881–887. [Google Scholar] [CrossRef]
- Kuter, D.J.; Macahilig, C.; Grotzinger, K.M.; Poston, S.A.; Wang, P.F.; Dawson, K.L.; Ward, M. Treatment patterns and clinical outcomes in patients with chronic immune thrombocytopenia (ITP) switched to eltrombopag or romiplostim. Int. J. Hematol. 2015, 101, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, S.; Carpenedo, M.; Mazzucconi, M.G.; De Stefano, V.; Carrai, V.; Ruggeri, M.; Specchia, G.; Vianelli, N.; Pane, F.; Consoli, U.; et al. Alternate use of thrombopoietin receptor agonists in adult primary immune thrombocytopenia patients: A retrospective collaborative survey from Italian hematology centers. Am. J. Hematol. 2018, 93, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Lozano, M.L.; Mingot-Castellano, M.E.; Perera, M.M.; Jarque, I.; Campos-Alvarez, R.M.; González-López, T.J.; Carreño-Tarragona, G.; Bermejo, N.; Lopez-Fernandez, M.F.; de Andrés, A.; et al. Deciphering predictive factors for choice of thrombopoietin receptor agonist, treatment free responses, and thrombotic events in immune thrombocytopenia. Sci. Rep. 2019, 9, 16680. [Google Scholar] [CrossRef] [PubMed]
- González-López, T.J.; Pascual, C.; Álvarez-Román, M.T.; Fernández-Fuertes, F.; Sánchez-González, B.; Caparrós, I.; Jarque, I.; Mingot-Castellano, M.E.; Hernández-Rivas, J.A.; Martín-Salces, M.; et al. Successful discontinuation of eltrombopag after complete remission in patients with primary immune thrombocytopenia. Am. J. Hematol. 2015, 90, E40–E43. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Fain, O.; Ebbo, M.; Roudot-Thoraval, F.; Limal, N.; Khellaf, M.; Schleinitz, N.; Bierling, P.; Languille, L.; Godeau, B.; et al. The temporary use of thrombopoietin-receptor agonists may induce a prolonged remission in adult chronic immune thrombocytopenia. Results of a French observational study. Br. J. Haematol. 2014, 165, 865–869. [Google Scholar] [CrossRef]
- Jurczak, W.; Chojnowski, K.; Mayer, J.; Krawczyk, K.; Jamieson, B.D.; Tian, W.; Allen, L.F. Phase 3 randomised study of avatrombopag, a novel thrombopoietin receptor agonist for the treatment of chronic immune thrombocytopenia. Br. J. Haematol. 2018, 183, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Doptelet Epar. Available online: https://www.ema.europa.eu/en/documents/product-information/doptelet-epar-product-information_en.pdf (accessed on 11 February 2024).
- Al-Samkari, H.; Jiang, D.; Gernsheimer, T.; Liebman, H.; Lee, S.; Wojdyla, M.; Vredenburg, M.; Cuker, A. Adults with immune thrombocytopenia who switched to avatrombopag following prior treatment with eltrombopag or romiplostim: A multicentre US study. Br. J. Haematol. 2022, 197, 359–366. [Google Scholar] [CrossRef]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Treliński, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef]
- Boccia, R.; Cooper, N.; Ghanima, W.; Boxer, M.A.; Hill, Q.A.; Sholzberg, M.; Tarantino, M.D.; Todd, L.K.; Tong, S.; Bussel, J.B.; et al. Fostamatinib is an effective second-line therapy in patients with immune thrombocytopenia. Br. J. Haematol. 2020, 190, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Tavlesse Epar. Available online: https://www.ema.europa.eu/en/documents/product-information/tavlesse-epar-product-information_en.pdf (accessed on 11 February 2024).
- Cooper, N.; Ghanima, W.; Hill, Q.A.; Nicolson, P.L.; Markovtsov, V.; Kessler, C. Recent advances in understanding spleen tyrosine kinase (SYK) in human biology and disease, with a focus on fostamatinib. Platelets 2023, 34, 2131751. [Google Scholar] [CrossRef]
- Martinez-Botia, P.; Meinders, M.; De Cuyper, I.M.; Eble, J.A.; Semple, J.W.; Gutierrez, L. Dissecting platelet proteomics to understand the pathophysiology of immune thrombocytopenia: Studies in mouse models. Blood Adv. 2022, 6, 3529–3534. [Google Scholar] [CrossRef]
- Lucchesi, A.; Fattizzo, B.; De Stefano, V.; Ruggeri, M.; Siragusa, S.; Vianelli, N.; Zaja, F.; Rodeghiero, F. Use and positioning of fostamatinib in the management of primary chronic immune thrombocytopenia: An Italian expert opinion. Ther. Adv. Hematol. 2023, 14, 20406207221147777. [Google Scholar] [CrossRef]
- Mabthera Epar. Available online: https://www.ema.europa.eu/en/documents/product-information/mabthera-epar-product-information_en.pdf (accessed on 12 February 2024).
- Lucchini, E.; Zaja, F.; Bussel, J. Rituximab in the treatment of immune thrombocytopenia: What is the role of this agent in 2019? Haematologica 2019, 104, 1124–1135. [Google Scholar] [CrossRef]
- Press, O.W.; Appelbaum, F.; Ledbetter, J.A.; Martin, P.J.; Zarling, J.; Kidd, P.; Thomas, E.D. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood 1987, 69, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kueck, B. Rituxan in the treatment of cold agglutinin disease. Blood 1998, 92, 3490–3491. [Google Scholar] [CrossRef] [PubMed]
- Shvidel, L.; Klepfish, A.; Berrebi, A. Successful treatment with Rituximab for relapsing immune thrombocytopenic purpura (ITP) associated with low-grade non-Hodgkin’s lymphoma. Am. J. Hematol. 2001, 67, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R.; Pagano, A.; Stipa, E.; Amadori, S. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adults with chronic idiopathic thrombocytopenic purpura. Blood 2001, 98, 952–957. [Google Scholar] [CrossRef]
- Wang, J.; Wiley, J.M.; Luddy, R.; Greenberg, J.; Feuerstein, M.A.; Bussel, J.B. Chronic immune thrombocytopenic purpura in children: Assessment of rituximab treatment. J. Pediatr. 2005, 146, 217–221. [Google Scholar] [CrossRef]
- Patel, V.L.; Mahévas, M.; Lee, S.Y.; Stasi, R.; Cunningham-Rundles, S.; Godeau, B.; Kanter, J.; Neufeld, E.; Taube, T.; Ramenghi, U.; et al. Outcomes 5 years after response to rituximab therapy in children and adults with immune thrombocytopenia. Blood 2012, 119, 5989–5995. [Google Scholar] [CrossRef]
- Dong, Y.; Yue, M.; Hu, M. The Efficacy and Safety of Different Dosages of Rituximab for Adults with Immune Thrombocytopenia: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2021, 2021, 9992086. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shi, Y.; He, Z.; Chen, Q.; Liu, Z.; Yu, L.; Wang, C. The efficacy and safety of low-dose rituximab in immune thrombocytopenia: A systematic review and meta-analysis. Platelets 2019, 30, 690–697. [Google Scholar] [CrossRef]
- Khellaf, M.; Charles-Nelson, A.; Fain, O.; Terriou, L.; Viallard, J.-F.; Cheze, S.; Graveleau, J.; Slama, B.; Audia, S.; Ebbo, M.; et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: Results from a prospective registry including 248 patients. Blood 2014, 124, 3228–3236. [Google Scholar] [CrossRef]
- Mahévas, M.; Ebbo, M.; Audia, S.; Bonnotte, B.; Schleinitz, N.; Durand, J.-M.; Chiche, L.; Khellaf, M.; Bierling, P.; Roudot-Thoraval, F.; et al. Efficacy and safety of rituximab given at 1,000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am. J. Hematol. 2013, 88, 858–861. [Google Scholar] [CrossRef]
- Zaja, F.; Baccarani, M.; Mazza, P.; Bocchia, M.; Gugliotta, L.; Zaccaria, A.; Vianelli, N.; Defina, M.; Tieghi, A.; Amadori, S.; et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010, 115, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- Gudbrandsdottir, S.; Birgens, H.S.; Frederiksen, H.; Jensen, B.A.; Jensen, M.K.; Kjeldsen, L.; Klausen, T.W.; Larsen, H.; Mourits-Andersen, H.T.; Nielsen, C.H.; et al. Rituximab and dexamethasone vs. dexamethasone monotherapy in newly diagnosed patients with primary immune thrombocytopenia. Blood 2013, 121, 1976–1981. [Google Scholar] [CrossRef]
- Bussel, J.B.; Lee, C.S.; Seery, C.; Imahiyerobo, A.A.; Thompson, M.V.; Catellier, D.; Turenne, I.G.; Patel, V.L.; Basciano, P.A.; Elstrom, R.L.; et al. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica 2014, 99, 1264–1271. [Google Scholar] [CrossRef]
- Chugh, S.; Darvish-Kazem, S.; Lim, W.; Crowther, M.A.; Ghanima, W.; Wang, G.; Heddle, N.M.; Kelton, J.G.; Arnold, D.M. Rituximab plus standard of care for treatment of primary immune thrombocytopenia: A systematic review and meta-analysis. Lancet Haematol. 2015, 2, e75–e81. [Google Scholar] [CrossRef] [PubMed]
- Kaznelson, P. Verschwinden der hamorrhagischen diathese bei einem falle von essentieller thrombopenie (frank) nach milzexstiparation: Splenogene thrombolytische purpura. Wien. Klin. Wochenschr. 1916, 29, 1451. [Google Scholar]
- Harrington, W.J.; Minnich, V.; Hollingsworth, J.W.; Moore, C.V. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J. Lab. Clin. Med. 1951, 38, 1–10. [Google Scholar] [PubMed]
- Palandri, F.; Polverelli, N.; Sollazzo, D.; Romano, M.; Catani, L.; Cavo, M.; Vianelli, N. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am. J. Hematol. 2016, 91, E267–E272. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Arnold, D.M.; McCrae, K.R. Splenectomy for immune thrombocytopenia: Down but not out. Blood 2018, 131, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Kojouri, K.; Vesely, S.K.; Terrell, D.R.; George, J.N. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: A systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004, 104, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.; White, R.H.; Brunson, A.; Wun, T. Splenectomy and the incidence of venous thromboembolism and sepsis in patients with immune thrombocytopenia. Blood 2013, 121, 4782–4790. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Porras, J.R.; Escalante, F.; Pardal, E.; Sierra, M.; Garcia-Frade, L.J.; Redondo, S.; Arefi, M.; Aguilar, C.; Ortega, F.; De Cabo, E.; et al. Safety and efficacy of splenectomy in over 65-yrs-old patients with immune thrombocytopenia. Eur. J. Haematol. 2013, 91, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Mageau, A.; Terriou, L.; Ebbo, M.; Souchaud-Debouverie, O.; Orvain, C.; Graveleau, J.; Lega, J.-C.; Ruivard, M.; Viallard, J.-F.; Cheze, S.; et al. Splenectomy for primary immune thrombocytopenia revisited in the era of thrombopoietin receptor agonists: New insights for an old treatment. Am. J. Hematol. 2022, 97, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Moulis, G.; Germain, J.; Comont, T.; Brun, N.; Dingremont, C.; Castel, B.; Arista, S.; Sailler, L.; Lapeyre-Mestre, M.; Beyne-Rauzy, O.; et al. Newly diagnosed immune thrombocytopenia adults: Clinical epidemiology, exposure to treatments, and evolution. Results of the CARMEN multicenter prospective cohort. Am. J. Hematol. 2017, 92, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Imbach, P.; Kühne, T.; Müller, D.; Berchtold, W.; Zimmerman, S.; Elalfy, M.; Buchanan, G.R. Childhood ITP: 12 months follow-up data from the prospective registry I of the Intercontinental Childhood ITP Study Group (ICIS). Pediatr. Blood Cancer 2006, 46, 351–356. [Google Scholar] [CrossRef]
- Miltiadous, O.; Hou, M.; Bussel, J.B. Identifying and treating refractory ITP: Difficulty in diagnosis and role of combination treatment. Blood 2020, 135, 472–490. [Google Scholar] [CrossRef]
- Vianelli, N.; Auteri, G.; Buccisano, F.; Carrai, V.; Baldacci, E.; Clissa, C.; Bartoletti, D.; Giuffrida, G.; Magro, D.; Rivolti, E.; et al. Refractory primary immune thrombocytopenia (ITP): Current clinical challenges and therapeutic perspectives. Ann. Hematol. 2022, 101, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.M.; Clerici, B.; Ilicheva, E.; Ghanima, W. Refractory immune thrombocytopenia in adults: Towards a new definition. Br. J. Haematol. 2023, 203, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Mahévas, M.; Gerfaud-Valentin, M.; Moulis, G.; Terriou, L.; Audia, S.; Guenin, S.; Le Guenno, G.; Salles, G.; Lambotte, O.; Limal, N.; et al. Characteristics, outcome, and response to therapy of multirefractory chronic immune thrombocytopenia. Blood 2016, 128, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Gudbrandsdottir, S.; Leven, E.; Imahiyerobo, A.; Lee, C.S.; Bussel, J. Combination of thrombopoietin receptor agonists, immunosuppressants and intravenous immunoglobulin as treatment of severe refractory immune thrombocytopenia in adults and children. Br. J. Haematol. 2020, 189, e37–e40. [Google Scholar] [CrossRef] [PubMed]
- Crickx, E.; Ebbo, M.; Rivière, E.; Souchaud-Debouverie, O.; Terriou, L.; Audia, S.; Ruivard, M.; Asli, B.; Marolleau, J.-P.; Méaux-Ruault, N.; et al. Combining thrombopoietin receptor agonists with immunosuppressive drugs in adult patients with multirefractory immune thrombocytopenia, an update on the French experience. Br. J. Haematol. 2023, 202, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Neufeld, E.J. Novel therapeutics and future directions for refractory immune thrombocytopenia. Br. J. Haematol. 2023, 203, 65–78. [Google Scholar] [CrossRef]
- Audia, S.; Bonnotte, B. Emerging Therapies in Immune Thrombocytopenia. J. Clin. Med. 2021, 10, 1004. [Google Scholar] [CrossRef]
- Kuter, D.J. Novel therapies for immune thrombocytopenia. Br. J. Haematol. 2022, 196, 1311–1328. [Google Scholar] [CrossRef] [PubMed]
- Provan, D.; Semple, J.W. Recent advances in the mechanisms and treatment of immune thrombocytopenia. EBioMedicine 2022, 76, 103820. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Kazmierczak, M.; Jarque, I.; Musteata, V.; Trelinski, J.; Cooper, N.; Kiessling, P.; Massow, U.; Woltering, F.; Snipes, R.; et al. Phase 2 multiple-dose study of an FcRn inhibitor, rozanolixizumab, in patients with primary immune thrombocytopenia. Blood Adv. 2020, 4, 4136–4146. [Google Scholar] [CrossRef]
- Broome, C.M.; McDonald, V.; Miyakawa, Y.; Carpenedo, M.; Kuter, D.J.; Al-Samkari, H.; Bussel, J.B.; Godar, M.; Ayguasanosa, J.; De Beuf, K.; et al. Efficacy and safety of the neonatal Fc receptor inhibitor efgartigimod in adults with primary immune thrombocytopenia (ADVANCE IV): A multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2023, 402, 1648–1659. [Google Scholar] [CrossRef] [PubMed]
- Kuter, D.J.; Efraim, M.; Mayer, J.; Trneny, M.; McDonald, V.; Bird, R.; Regenbogen, T.; Garg, M.; Kaplan, Z.; Tzvetkov, N.; et al. Rilzabrutinib, an Oral BTK Inhibitor, in Immune Thrombocytopenia. N. Engl. J. Med. 2022, 386, 1421–1431. [Google Scholar] [CrossRef]
- Broome, C.M.; Roth, A.; Kuter, D.J.; Scully, M.; Smith, R.; Wang, J.; Reuter, C.; Hobbs, W.; Daak, A. Safety and efficacy of classical complement pathway inhibition with sutimlimab in chronic immune thrombocytopenia. Blood Adv. 2023, 7, 987–996. [Google Scholar] [CrossRef]
- Alvarez-Roman, M.T.; Rivas Pollmar, M.I.; Bernardino, J.I.; Lozano, M.L.; Martin-Salces, M.; Fernandez-Bello, I.; Jimenez-Yuste, V.; Butta, N.V. Thrombopoietin receptor agonists in conjunction with oseltamivir for immune thrombocytopenia. AIDS 2016, 30, 1141–1142. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Wu, Y.; Zhou, H.; Qin, P.; Ni, H.; Peng, J.; Hou, M. Successful treatment with oseltamivir phosphate in a patient with chronic immune thrombocytopenia positive for anti-GPIb/IX autoantibody. Platelets 2015, 26, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, J.; Shao, L.; Yuan, C.; Zhao, H.; Li, D.; Wang, Z.; Han, P.; Yu, Y.; Xu, M.; et al. Dexamethasone plus oseltamivir versus dexamethasone in treatment-naive primary immune thrombocytopenia: A multicentre, randomised, open-label, phase 2 trial. Lancet Haematol. 2021, 8, e289–e298. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yi, Z.; Wang, S.; Li, Z. The effect of decitabine on megakaryocyte maturation and platelet release. Thromb. Haemost. 2011, 106, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hou, Y.; Liu, X.; Qiu, J.; Feng, Q.; Wang, Y.; Zhang, X.; Min, Y.; Shao, L.; Liu, X.; et al. Low-dose decitabine promotes megakaryocyte maturation and platelet production in healthy controls and immune thrombocytopenia. Thromb. Haemost. 2015, 113, 1021–1034. [Google Scholar] [CrossRef]
- Zhou, H.; Qin, P.; Liu, Q.; Yuan, C.; Hao, Y.; Zhang, H.; Wang, Z.; Ran, X.; Chu, X.; Yu, W.; et al. A prospective, multicenter study of low dose decitabine in adult patients with refractory immune thrombocytopenia. Am. J. Hematol. 2019, 94, 1374–1381. [Google Scholar] [CrossRef]

{kind=link}
| Year | Therapy |
|---|---|
| 1735 | Elixirium acidum halleri (citric acid) |
| 19th century | Moderate exercise in the open air, a generous diet, and the free use of wine Purgatives |
| 1916 | First splenectomyBeginning of the splenectomy era |
| 1951 | CorticosteroidsBeginning of the immunosuppressant era |
| 1972 | First randomized double-blind study of corticosteroids versus placebo |
| 1981 | Intravenous immunoglobulins |
| 1983 | Specific anti-D immunoglobulin |
| 1994 | Dexamethasone in ITP was described for the first time |
| 2001 | First prospective study of rituximab use in chronic ITP |
| 2008 | FDA approval of romiplostim and eltrombopagBeginning of the TPO-RA era |
| 2018 | FDA approval of fostamatinib |
| 2019 | FDA approval of avatrombopag |
| 2024 | New drugs in clinical development |
| TPO-RA | Fostamatinib | Rituximab | Splenectomy | |
|---|---|---|---|---|
| Recommendation | First strategy | Second strategy | Second strategy | Second strategy |
| Advantages | Good responses (70–95% initial, 40–60% sustained) Possibility of discontinuation Safe therapies | No risk of thrombosis or immunosuppression | Good tolerance No chronic medication No risk of thrombosis | Good responses (>80% initial, 50–75% sustained) No chronic medication Low cost |
| Disadvantages | Cost | Low sustained responses (18% after 24 weeks) Adverse effects (gastrointestinal effects, hypertension) Cost | Low sustained responses (20% after 5 years) Immunosuppression | Adverse effects (surgical effects, thrombosis, infections) Need for vaccination |
| Description | Drug | Mechanism of Action |
|---|---|---|
| Recently approved treatments | ||
| TPO-RA | Avatrombopag | Stimulates the proliferation of megakaryocytes, maturation, and production of platelets |
| Syk inhibitor | Fostamatinib | Decreases splenic platelet clearance |
| Treatments in clinical development | ||
| FcRn inhibitors | Rozanolixizumab Efgartigimod | Decreases plasma levels of IgG (normal and pathological) |
| BTK inhibitor | Rilzabrutinib | Decreases splenic platelet clearance Decreases the production of autoantibodies |
| Anti-CD38 | Daratumumab Mezagitamab | Decreases the production of autoantibodies |
| Complement (C1s) inhibitor | Sutimlimab | Decreases complement-dependent cytotoxicity, thereby reducing platelet destruction |
| Neuraminidase inhibitor | Oseltamivir | Inhibits platelet desialylation, thus decreasing hepatic platelet clearance |
| Epigenetic modulation | Decitabine | Promotes the maturation of megakaryocytes and platelet production Enhances Treg lymphocytes and decreases proinflammatory cytokines |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Carballeira, D.; Bernardo, Á.; Caro, A.; Soto, I.; Gutiérrez, L. Treatment of Immune Thrombocytopenia: Contextualization from a Historical Perspective. Hematol. Rep. 2024, 16, 390-412. https://doi.org/10.3390/hematolrep16030039
Martínez-Carballeira D, Bernardo Á, Caro A, Soto I, Gutiérrez L. Treatment of Immune Thrombocytopenia: Contextualization from a Historical Perspective. Hematology Reports. 2024; 16(3):390-412. https://doi.org/10.3390/hematolrep16030039
Chicago/Turabian StyleMartínez-Carballeira, Daniel, Ángel Bernardo, Alberto Caro, Inmaculada Soto, and Laura Gutiérrez. 2024. "Treatment of Immune Thrombocytopenia: Contextualization from a Historical Perspective" Hematology Reports 16, no. 3: 390-412. https://doi.org/10.3390/hematolrep16030039
APA StyleMartínez-Carballeira, D., Bernardo, Á., Caro, A., Soto, I., & Gutiérrez, L. (2024). Treatment of Immune Thrombocytopenia: Contextualization from a Historical Perspective. Hematology Reports, 16(3), 390-412. https://doi.org/10.3390/hematolrep16030039