
Genetics of Inner Ear Malformations: A Review

 , , , and
, , , and
Abstract
:1. Introduction
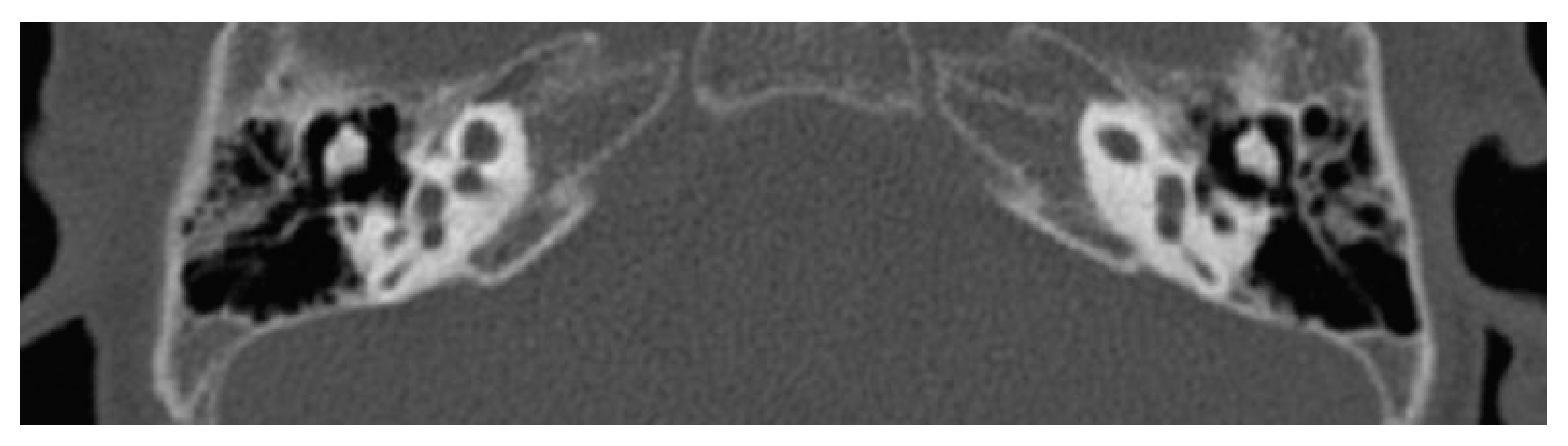
2. Complete Labyrinthine Aplasia
3. Rudimentary Otocyst
4. Common Cavity
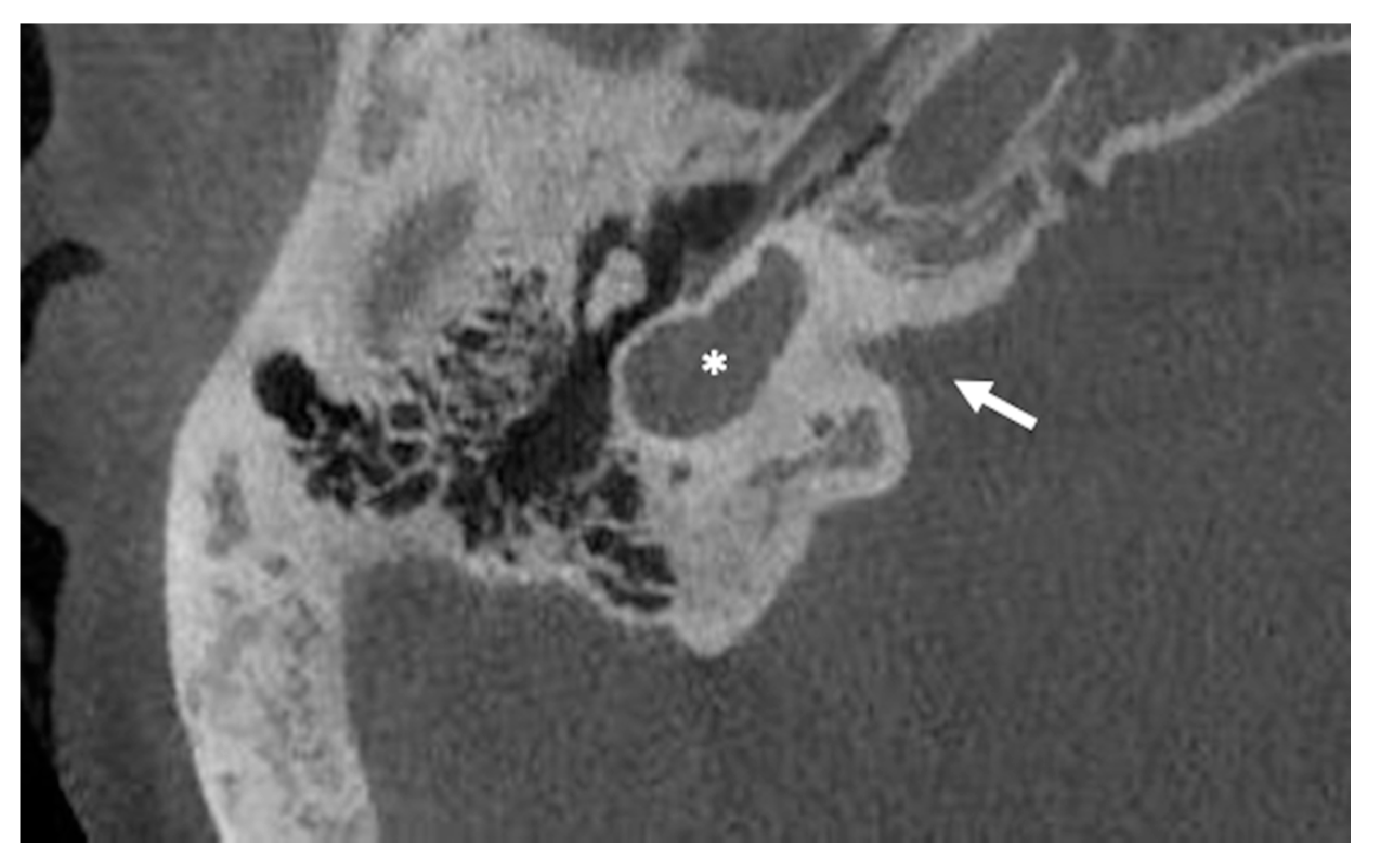
5. Cochlear Aplasia (with and without a Dilated Vestibule)
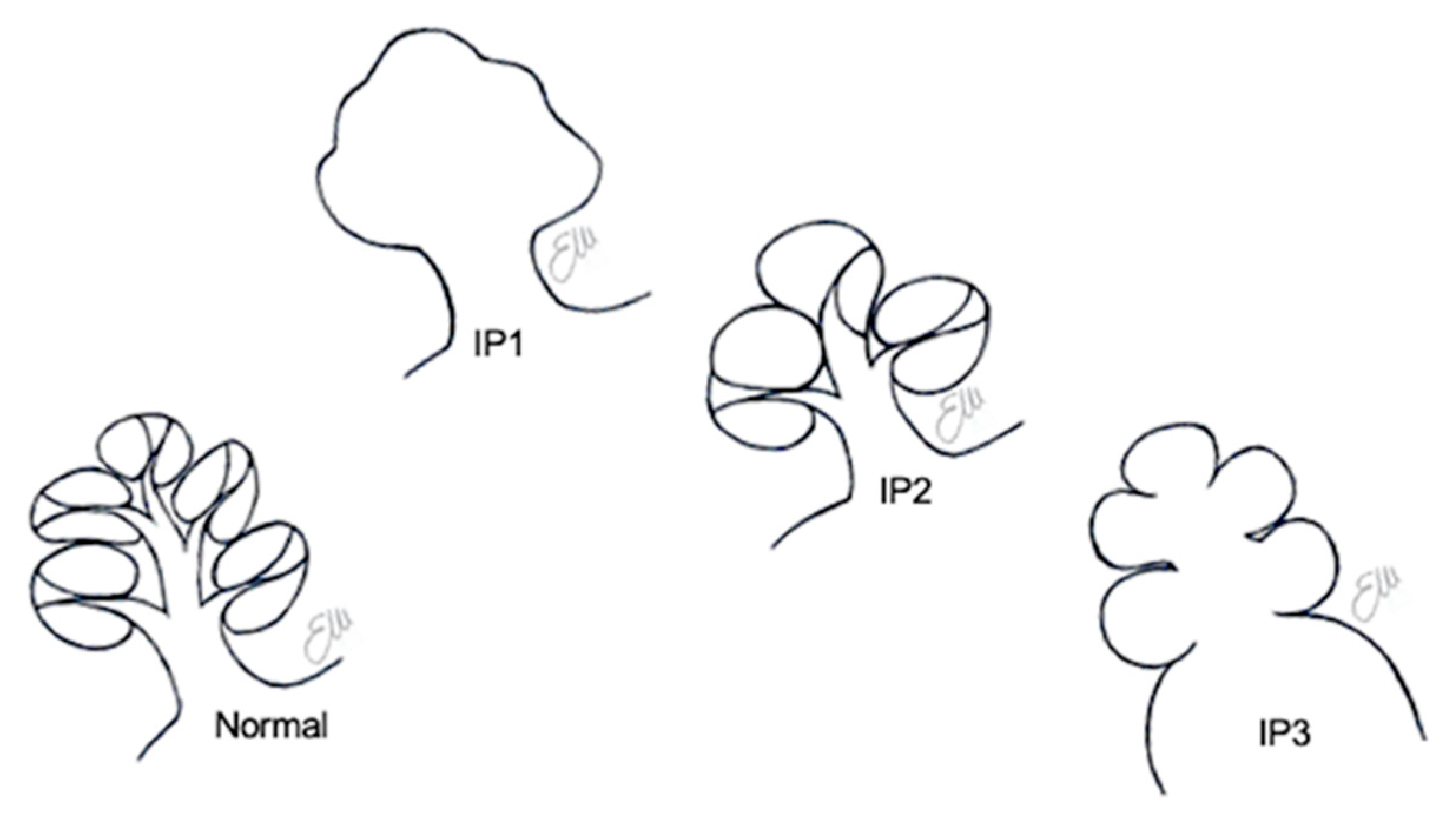
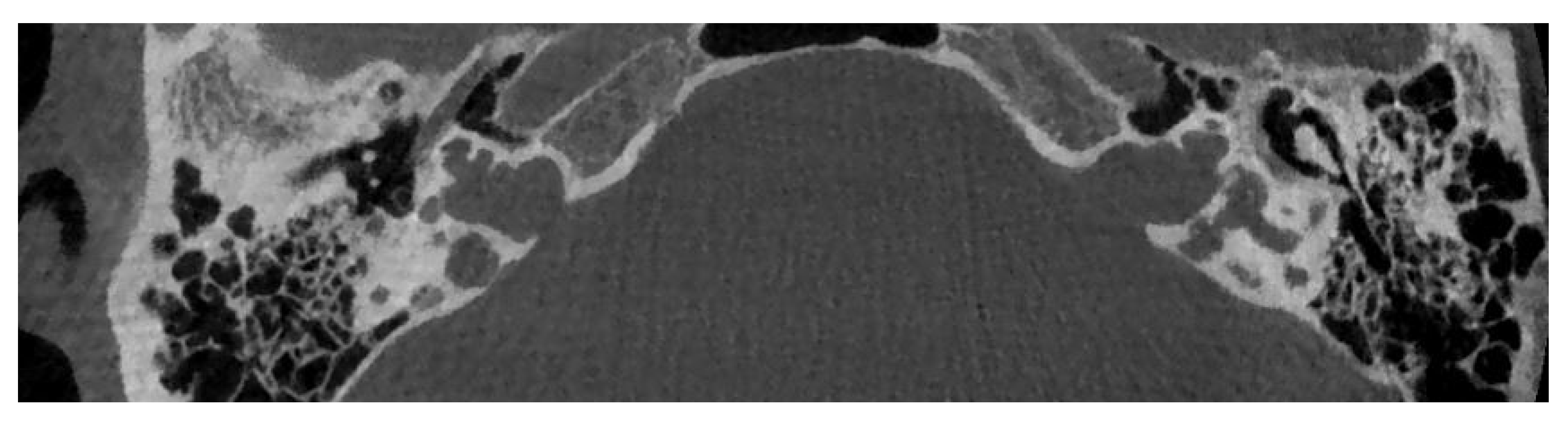
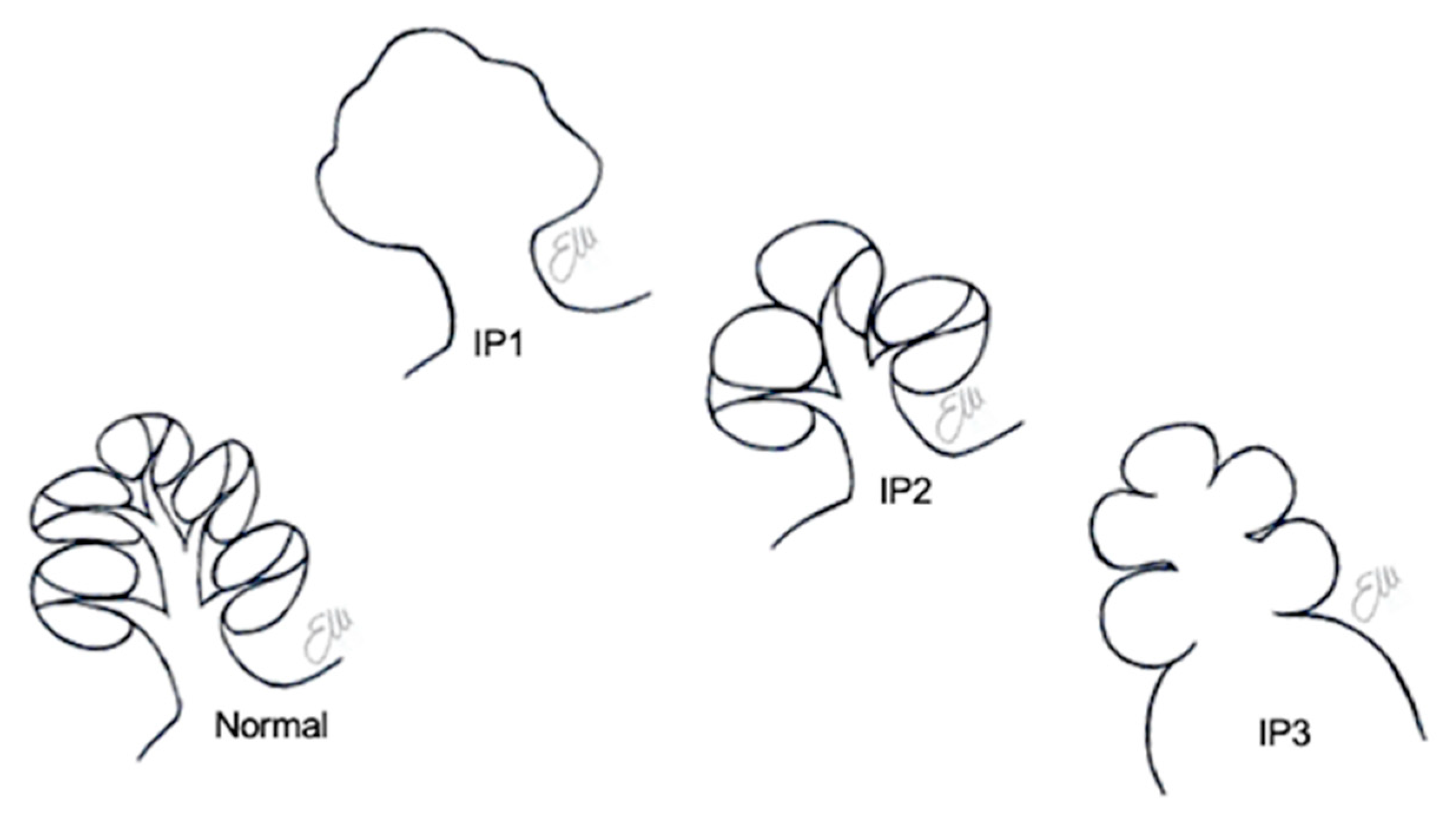
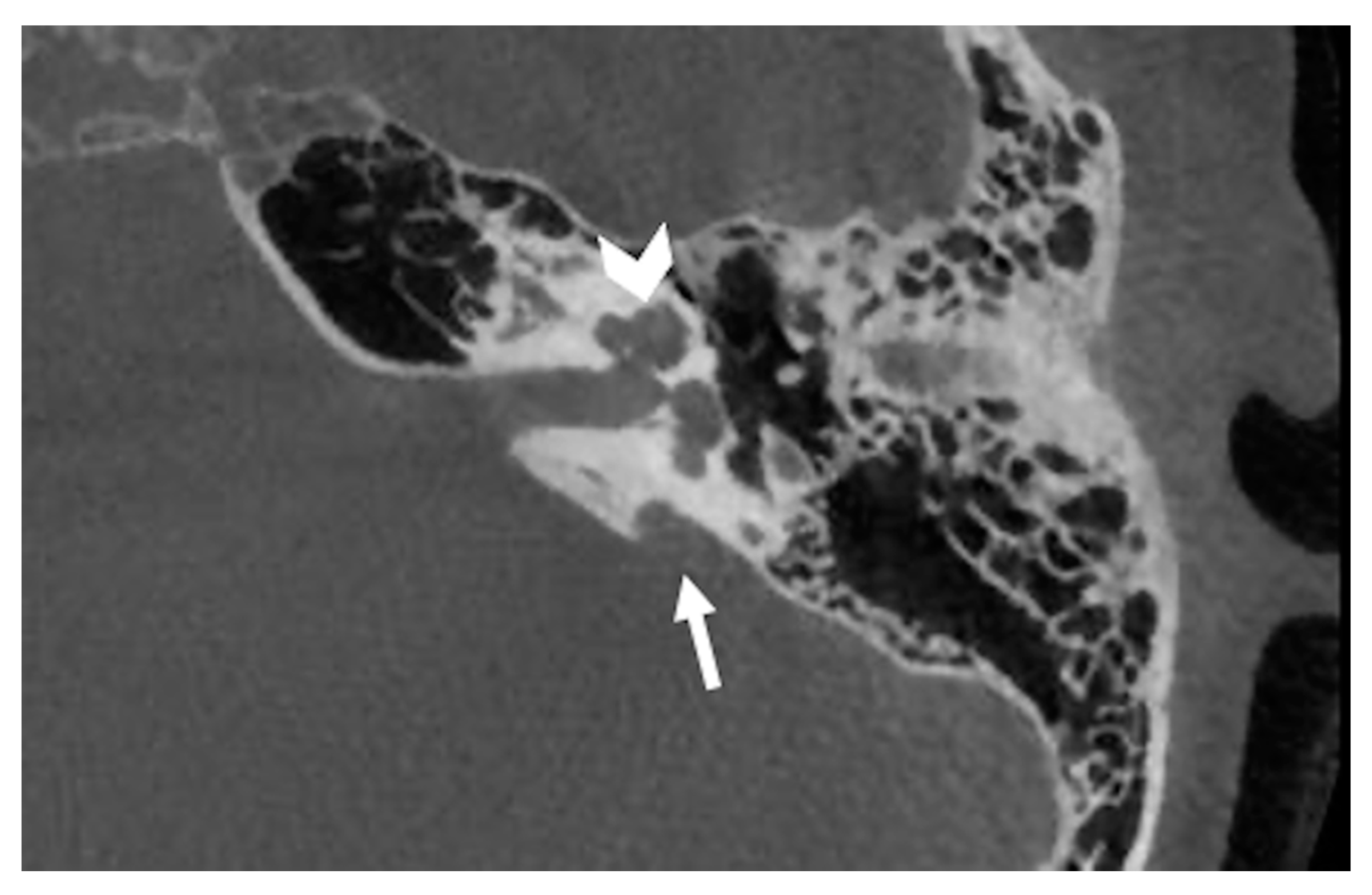
6. Incomplete Partitions
6.1. Incomplete Partition Type 1
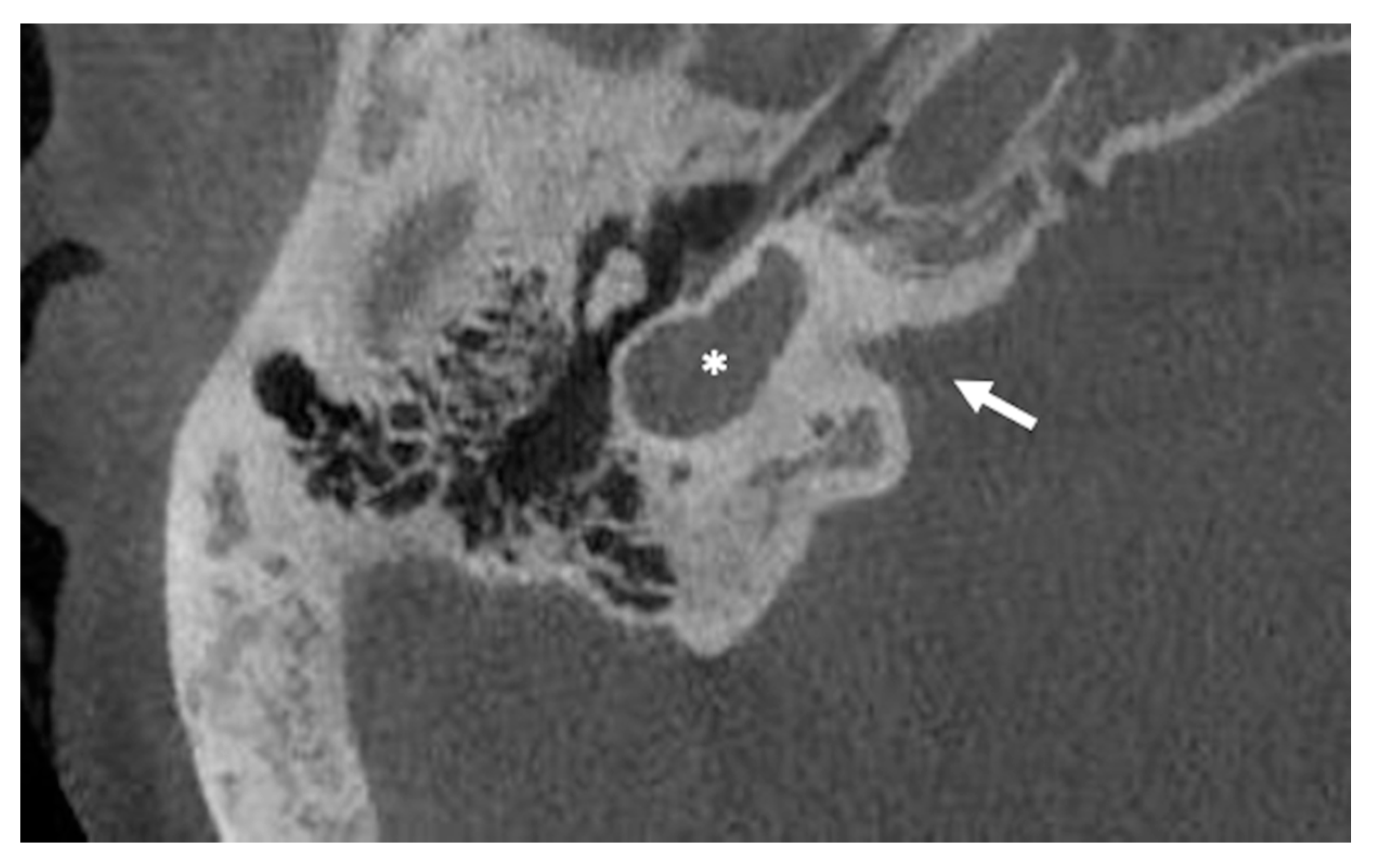
6.2. Incomplete Partition Type 2
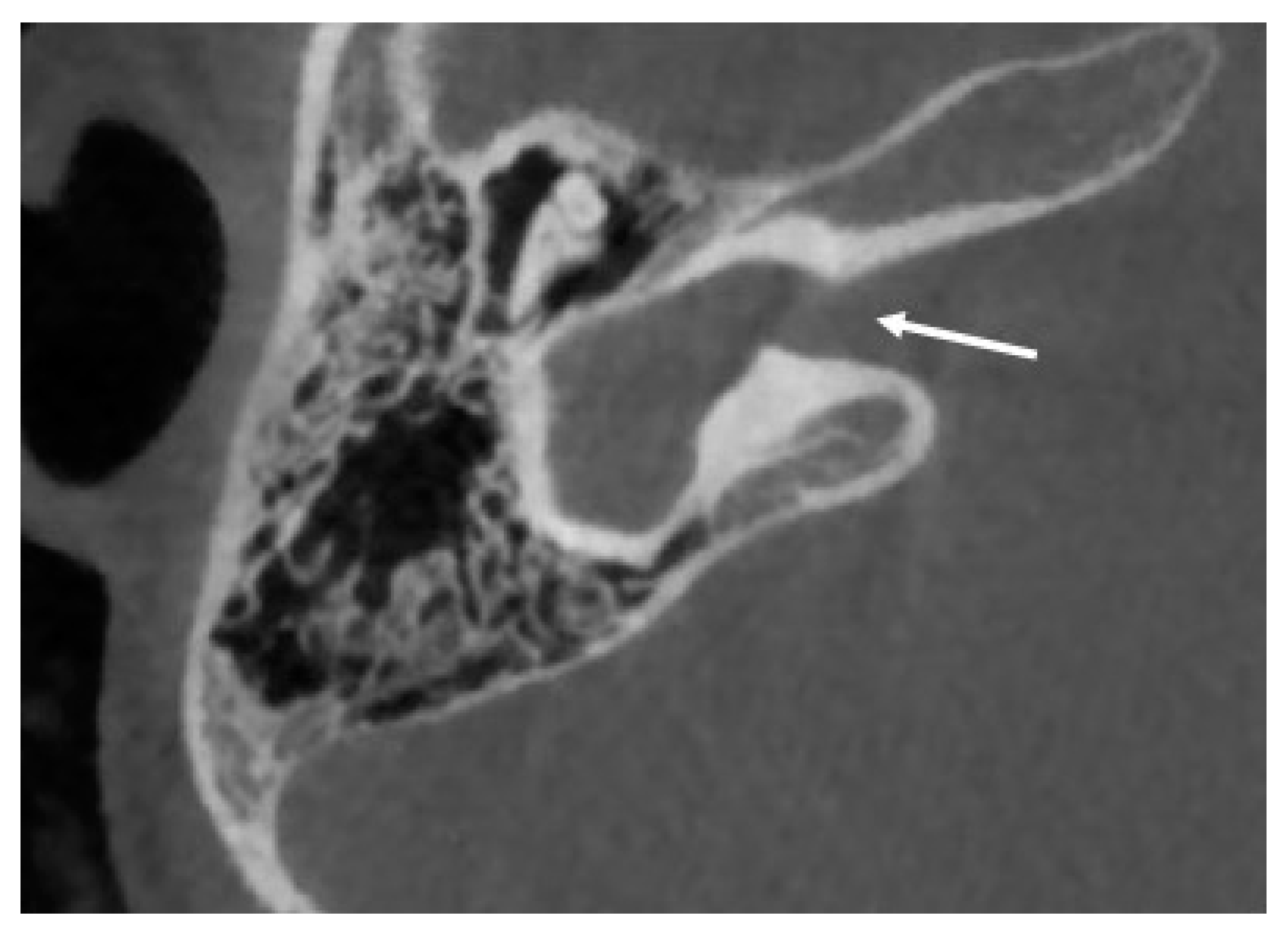
6.3. Incomplete Partition Type 3
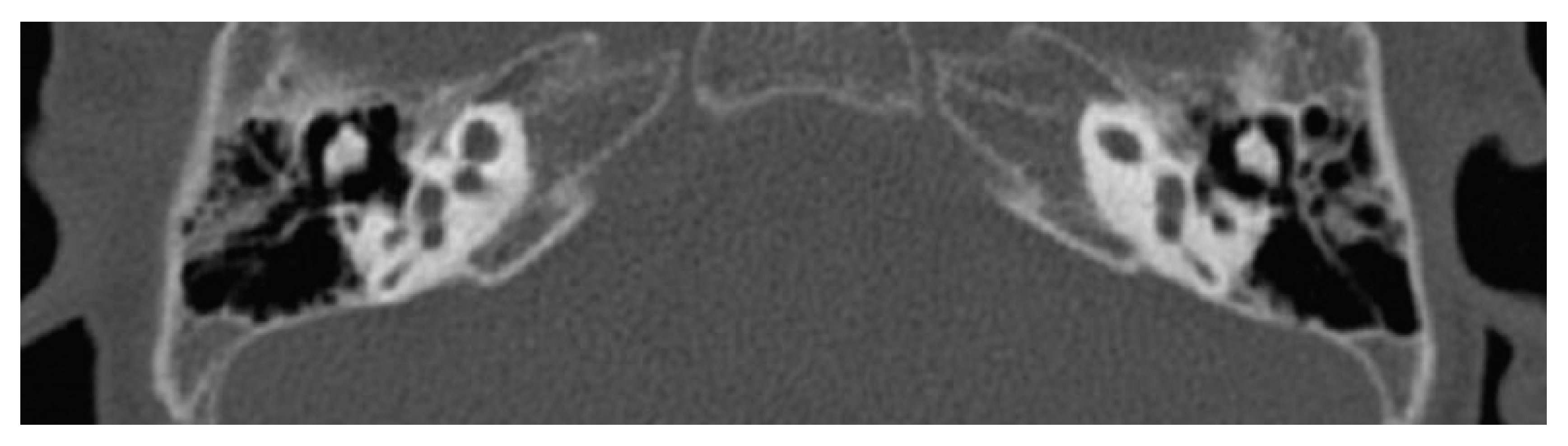
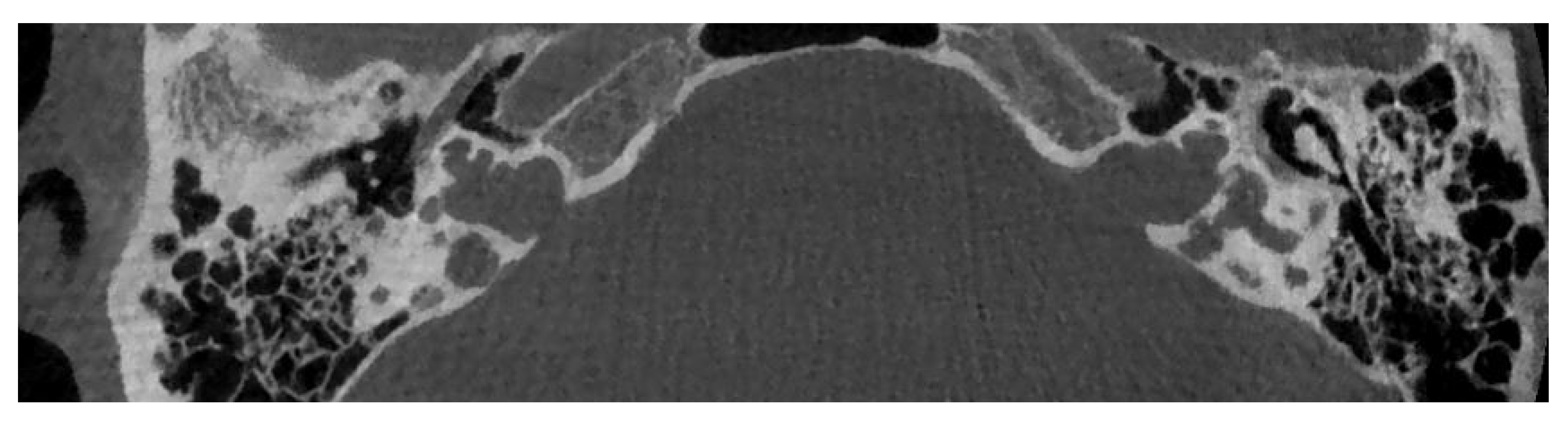
7. Cochlear Hypoplasia
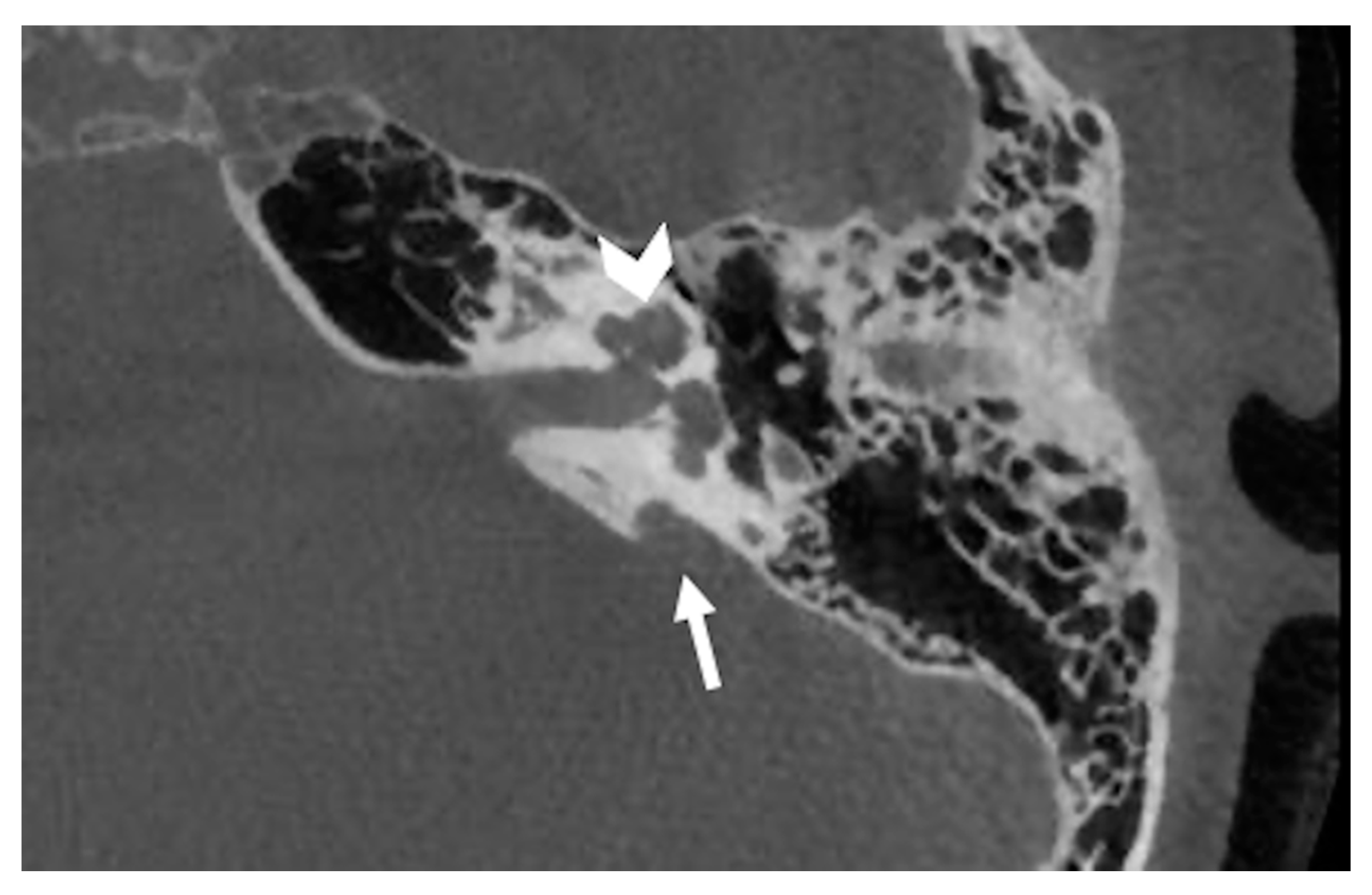
8. Posterior Labyrinth Anomalies
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sennaroğlu, L.; Bajin, M.D. Classification and Current Management of Inner Ear Malformations. Balk. Med. J. 2017, 34, 397–411. [Google Scholar] [CrossRef]
- Brotto, D.; Uberti, A.; Manara, R. From Mondini to the latest inner ear malformations’ classifications: An historical and critical review. Hear. Balance Commun. 2019, 17, 241–248. [Google Scholar] [CrossRef]
- Sennaroglu, L.; Saatci, I. A new classification for cochleovestibular malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Sennaroğlu, L.; Yarali, M.; Sennaroğlu, G.; Çinar, B.Ç.; Batuk, M.Ö.; Yücel, E.; Bilginer, B.; Bajin, M.D.; Winter, M.; Wilkinson, E.P. Simultaneous Cochlear and Auditory Brainstem Implantation in Children With Severe Inner Ear Malformations: Initial Surgical and Audiological Results. Otol. Neurotol. 2020, 41, 625–630. [Google Scholar] [CrossRef]
- Sennaroğlu, L.; Sennaroğlu, G.; Yücel, E.; Bilginer, B.; Atay, G.; Bajin, M.D.; Mocan, B.Ö.; Yaral, M.; Aslan, F.; Çnar, B.Ç.; et al. Long-term Results of ABI in Children With Severe Inner Ear Malformations. Otol. Neurotol. 2016, 37, 865–872. [Google Scholar] [CrossRef]
- Sennaroğlu, L.; Bajin, M.D.; Çınar, B.Ç.; Yaralı, M.; Özbal Batuk, M. Stapedotomy in Congenital Fixation with Cochlear Hypoplasia: A New Concept among the Treatment Options for Cochlear Hypoplasia. J. Int. Adv. Otol. 2021, 17, 228–233. [Google Scholar] [CrossRef]
- Brotto, D.; Avato, I.; Lovo, E.; Muraro, E.; Bovo, R.; Trevisi, P.; Martini, A.; Manara, R. Epidemiologic, Imaging, Audiologic, Clinical, Surgical, and Prognostic Issues in Common Cavity Deformity: A Narrative Review. JAMA Otolaryngol. Neck Surg. 2019, 145, 72–78. [Google Scholar] [CrossRef]
- Jackler, R.K.; Luxford, W.M.; House, W.F. Congenital malformations of the inner ear: A classification based on embryogenesis. Laryngoscope 1987, 97, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Ozgen, B.; Oguz, K.K.; Atas, A.; Sennaroglu, L. Complete labyrinthine aplasia: Clinical and radiologic findings with review of the literature. Am. J. Neuroradiol. 2009, 30, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Michel, E.M. Mémoires sur les anomalies congénitales de l’oreille interne, avec la première observation authentique d’absence complète d’oreilles internes et de nerfs auditifs, et de l’absence partielle de l’oreille moyenne chez un sourd et muet de naissance, mort à l’âge de onze ans. Gaz. Med. Strasbourg. 1863, 4, 55–58. [Google Scholar]
- Daneshi, A.; Farhadi, M.; Asghari, A.; Emamjomeh, H.; Abbasalipour, P.; Hasanzadeh, S. Three familial cases of Michel’s aplasia. Otol. Neurotol. 2002, 23. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, S.F.; Tubergen, L.B. Unilateral complete aplasia of the inner ear with associated tracheoesophageal fistula: Report of a case. Otolaryngol. Neck Surg. 1979, 87, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Keeney, G.; Gebarski, S.S.; Brunberg, J.A. CT of severe inner ear anomalies, including aplasia, in a case of Wildervanck syndrome. AJNR Am. J. Neuroradiol. 1992, 13, 201–202. [Google Scholar] [PubMed]
- Tekin, M.; Hişmi, B.O.; Fitoz, S.; Ozdağ, H.; Cengiz, F.B.; Sirmaci, A.; Aslan, I.; Inceoğlu, B.; Yüksel-Konuk, E.B.; Yilmaz, S.T.; et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am. J. Hum. Genet. 2007, 80, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sensi, A.; Ceruti, S.; Trevisi, P.; Gualandi, F.; Busi, M.; Donati, I.; Neri, M.; Ferlini, A.; Martini, A. LAMM syndrome with middle ear dysplasia associated with compound heterozygosity for FGF3 mutations. Am. J. Med. Genet. Part A 2011, 155, 1096–1101. [Google Scholar] [CrossRef]
- Riazuddin, S.; Ahmed, Z.M.; Hegde, R.S.; Khan, S.N.; Nasir, I.; Shaukat, U.; Riazuddin, S.; Butman, J.A.; Griffith, A.J.; Friedman, T.B.; et al. Variable expressivity of FGF3 mutations associated with deafness and LAMM syndrome. BMC Med. Genet. 2011, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Al Yassin, A.; D’Arco, F.; Morín, M.; Pagarkar, W.; Harrop-Griffiths, K.; Shaida, A.; Fernández, E.; Cullup, T.; De-Souza, B.; Moreno-Pelayo, M.A.; et al. Three New Mutations and Mild, Asymmetrical Phenotype in the Highly Distinctive LAMM Syndrome: A Report of Eight Further Cases. Genes 2019, 10, 529. [Google Scholar] [CrossRef] [Green Version]
- Ordonez, J.; Tekin, M. Congenital Deafness with Labyrinthine Aplasia, Microtia, and Microdontia. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2020. [Google Scholar]
- Lallar, M.; Arora, V.; Saxena, R.; Puri, R.D.; Verma, I.C. Complete Labyrinthine Aplasia: A Unique Sign for Targeted Genetic Testing in Hearing Loss. J. Pediatr. Genet. 2021, 10, 070–073. [Google Scholar] [CrossRef]
- Higley, M.J.; Walkiewicz, T.W.; Miller, J.H.; Curran, J.G.; Towbin, R.B. Bilateral complete labyrinthine aplasia with bilateral internal carotid artery aplasia, developmental delay, and gaze abnormalities: A presumptive case of a rare HOXA1 mutation syndrome. Am. J. Neuroradiol. 2010, 32, E23–E25. [Google Scholar] [CrossRef] [Green Version]
- Giesemann, A.M.; Goetz, F.; Neuburger, J.; Lenarz, T.; Lanfermann, H. From labyrinthine aplasia to otocyst deformity. Neuroradiology 2009, 52, 147–154. [Google Scholar] [CrossRef]
- Bosley, T.M.; Salih, M.A.; Alorainy, I.A.; Oystreck, D.T.; Nester, M.; Abu-Amero, K.K.; Tischfield, M.A.; Engle, E.C. Clinical characterization of the HOXA1 syndrome BSAS variant. Neurology 2007, 69, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Horta, O.; Abad, C.; Sennaroglu, L.; Foster, J., 2nd; DeSmidt, A.; Bademci, G.; Tokgoz-Yilmaz, S.; Duman, D.; Cengiz, F.B.; Grati, M.; et al. ROR1 is essential for proper innervation of auditory hair cells and hearing in humans and mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5993–5998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loos, E.; Verhaert, N.; Willaert, A.; Devriendt, K.; Swillen, A.; Hermans, R.; Op de Beeck, K.; Hens, G. Malformations of the middle and inner ear on CT imaging in 22q11 deletion syndrome. Am. J. Med Genet. Part A 2016, 170, 2975–2983. [Google Scholar] [CrossRef]
- Joshi, V.M.; Navlekar, S.K.; Kishore, G.R.; Reddy, K.J.; Kumar, E.C. CT and MR imaging of the inner ear and brain in children with congenital sensorineural hearing loss. Radiographics 2012, 32, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.; de Caprona, D.; Busslinger, M.; Xu, P.; Fritzsch, B. Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev. Biol. 2010, 10, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nance, W.E.; Setleff, R.; McLeod, A.; Sweeney, A.; Cooper, C.; McConnell, F. X-linked mixed deafness with congenital fixation of the stapedial footplate and perilymphatic gusher. Birth. Defects Orig. Artic. Ser. 1971, 7, 64–69. [Google Scholar]
- Mori, T.; Westerberg, B.D.; Atashband, S.; Kozak, F.K. Natural history of hearing loss in children with enlarged vestibular aqueduct syndrome. J. Otolaryngol. Head Neck Surg. 2008, 37, 112–118. [Google Scholar]
- D’Arco, F.; Youssef, A.; Ioannidou, E.; Bisdas, S.; Pinelli, L.; Caro-Dominguez, P.; Nash, R.; Siddiqui, A.; Talenti, G. Temporal bone and intracranial abnormalities in syndromic causes of hearing loss: An updated guide. Eur. J. Radiol. 2020, 123, 108803. [Google Scholar] [CrossRef] [Green Version]
- Wémeau, J.L.; Kopp, P. Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 213–224. [Google Scholar] [CrossRef]
- Da Silva Costa, S.M.; Ramos, P.Z.; Arrojo Martins, F.T.; Sartorato, E.L. Genetic Diagnosis of Deafness. In The Role of Pendrin in Health and Disease; Dossena, S., Paulmichl, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 61–83. [Google Scholar]
- Roesch, S.; Rasp, G.; Sarikas, A.; Dossena, S. Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review. Audiol. Res. 2021, 11, 423–442. [Google Scholar] [CrossRef]
- Hu, X.; Liang, F.; Zhao, M.; Gong, A.; Berry, E.R.; Shi, Y.; Wang, Y.; Chen, Y.; Liu, A.; Qu, C. Mutational analysis of the SLC26A4 gene in Chinese sporadic nonsyndromic hearing-impaired children. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Qing, J.; Zhou, Y.; Lai, R.; Hu, P.; Ding, Y.; Wu, W.; Xiao, Z.; Ho, P.T.; Liu, Y.; Liu, J.; et al. Prevalence of mutations in GJB2, SLC26A4, and mtDNA in children with severe or profound sensorineural hearing loss in southwestern China. Genet. Test. Mol. Biomark. 2015, 19, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, X.; Chai, Y.; Chen, P.; He, L.; Wang, X.; Wu, H.; Yang, T. Mono-allelic mutations of SLC26A4 is over-presented in deaf patients with non-syndromic enlarged vestibular aqueduct. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1351–1353. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, H.; Liu, D.; Zhang, J.; Yang, Z.; Zhang, S.; Liu, H.; Li, R.; Tian, Y.; Zeng, B.; et al. Increased diagnosis of enlarged vestibular aqueduct by multiplex PCR enrichment and next-generation sequencing of the SLC26A4 gene. Mol. Genet. Genom. Med. 2021, 9, e1734. [Google Scholar] [CrossRef]
- Lin, Y.H.; Wu, C.C.; Lin, Y.H.; Lu, Y.C.; Chen, C.S.; Liu, T.C.; Chen, P.L.; Hsu, C.J. Targeted Next-Generation Sequencing Facilitates Genetic Diagnosis and Provides Novel Pathogenetic Insights into Deafness with Enlarged Vestibular Aqueduct. J. Mol. Diagn. 2019, 21, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.W.; Lee, S.C.; Lee, H.K.; Park, H.J. Genetic Screening of GJB2 and SLC26A4 in Korean Cochlear Implantees: Experience of Soree Ear Clinic. Clin. Exp. Otorhinolaryngol. 2012, 5, S10–S13. [Google Scholar] [CrossRef]
- Rah, Y.C.; Kim, A.R.; Koo, J.W.; Lee, J.H.; Oh, S.H.; Choi, B.Y. Audiologic presentation of enlargement of the vestibular aqueduct according to the SLC26A4 genotypes. Laryngoscope 2015, 125, E216–E222. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Nishio, S.Y.; Usami, S. Deafness Gene Study Consortium. Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: A large cohort study. J. Hum. Genet. 2014, 59, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, S.; Bernardinelli, E.; Nofziger, C.; Tóth, M.; Patsch, W.; Rasp, G.; Paulmichl, M.; Dossena, S. Functional Testing of SLC26A4 Variants-Clinical and Molecular Analysis of a Cohort with Enlarged Vestibular Aqueduct from Austria. Int. J. Mol. Sci. 2018, 19, 209. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.; Muskett, J.A.; King, K.A.; Zalewski, C.K.; Chattaraj, P.; Butman, J.A.; Kenna, M.A.; Chien, W.W.; Brewer, C.C.; Griffith, A.J. Hearing loss associated with enlarged vestibular aqueduct and zero or one mutant allele of SLC26A4. Laryngoscope 2017, 127, E238–E243. [Google Scholar] [CrossRef]
- Rehman, A.U.; Friedman, T.B.; Griffith, A.J. Unresolved questions regarding human hereditary deafness. Oral Dis. 2017, 23, 551–558. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Feng, Y.; He, C.; Liu, D.; Cai, X.; Jiang, L.; Chen, H.; Liu, C.; Wu, H.; et al. A New Genetic Diagnostic for Enlarged Vestibular Aqueduct Based on Next-Generation Sequencing. PLoS ONE 2016, 11, e0168508. [Google Scholar] [CrossRef] [Green Version]
- Sennaroğlu, L.; Bajin, M.D. Incomplete partition type III: A rare and difficult cochlear implant surgical indication. Auris Nasus Larynx 2018, 45, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Xiao, Y.; Zhang, F.; Luo, J.; Wang, R.; Liu, W.; Wang, H.; Xu, L. Cochlear Implantation in a Patient with a Novel POU3F4 Mutation and Incomplete Partition Type-III Malformation. Neural Plast. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sennaroglu, L. Histopathology of inner ear malformations: Do we have enough evidence to explain pathophysiology? Cochlea Implant. Int. 2015, 17, 3–20. [Google Scholar] [CrossRef] [PubMed]
- de Kok, Y.J.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.H.; Cremers, F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995, 267, 685–688. [Google Scholar] [CrossRef] [Green Version]
- Mathis, J.; Simmons, D.; He, X.; Swanson, L.; Rosenfeld, M. Brain 4: A novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J. 1992, 11, 2551–2561. [Google Scholar] [CrossRef]
- Siddiqui, A.; D’Amico, A.; Colafati, G.S.; Cicala, D.; Talenti, G.; Rajput, K.; Pinelli, L.; D’Arco, F. Hypothalamic malformations in patients with X-linked deafness and incomplete partition type 3. Neuroradiology 2019, 61, 949–952. [Google Scholar] [CrossRef]
- D’Arco, F.; Sanverdi, E.; O’Brien, W.T.; Taranath, A.; Talenti, G.; Blaser, S.I. The link between inner ear malformations and the rest of the body: What we know so far about genetic, imaging and histology. Neuroradiology 2020, 62, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Cinar, B.C.; Batuk, M.O.; Tahir, E.; Sennaroglu, G.; Sennaroglu, L. Audiologic and radiologic findings in cochlear hypoplasia. Auris Nasus Larynx 2017, 44, 655–663. [Google Scholar] [CrossRef]
- Talenti, G.; Manara, R.; Brotto, D.; D’Arco, F. High-resolution 3 T magnetic resonance findings in cochlear hypoplasias and incomplete partition anomalies: A pictorial essay. Br. J. Radiol. 2018, 91, 2018120. [Google Scholar] [CrossRef]
- Chen, A.; Francis, M.; Ni, L.; Cremers, C.W.; Kimberling, W.J.; Sato, Y.; Phelps, P.D.; Bellman, S.C.; Wagner, M.J.; Pembrey, M.; et al. Phenotypic manifestations of branchio-oto-renal syndrome. Am. J. Med. Genet. 1995, 58, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Kemperman, M.H.; Stinckens, C.; Kumar, S.; Huygen, P.L.; Joosten, F.B.; Cremers, C.W. Progressive fluctuant hearing loss, enlarged vestibular aqueduct, and cochlear hypoplasia in branchio-oto-renal syndrome. Otol. Neurotol. 2001, 22, 637–643. [Google Scholar] [CrossRef]
- Davide, B.; Renzo, M.; Sara, G.; Elisa, L.; Rodica, M.; Irene, T.; Alessandro, C.; Giovanni, S.; Valentina, S.; Roberto, B.; et al. Oculo-auriculo-vertebral spectrum: Going beyond the first and second pharyngeal arch involvement. Neuroradiology 2017, 59, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Kari, E.; Llaci, L.; Go, J.L.; Naymik, M.; Knowles, J.A.; Leal, S.M.; Rangasamy, S.; Huentelman, M.J.; Friedman, R.A.; Schrauwen, I. A de novo SIX1 variant in a patient with a rare nonsyndromic cochleovestibular nerve abnormality, cochlear hypoplasia, and bilateral sensorineural hearing loss. Mol. Genet. Genom. Med. 2019, 7, e995. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Gao, X.; Su, Y.; Han, M.; Gao, B.; Guo, C.; Kang, D.; Huang, S.; Yuan, Y.; et al. Analysis of genotype-phenotype relationships in 90 Chinese probands with Waardenburg syndrome. Hum. Genet. 2021, 1–14. [Google Scholar] [CrossRef]
- Oysu, C.; Oysu, A.; Aslan, I.; Tinaz, M. Temporal bone imaging findings in Waardenburg’s syndrome. Int. J. Pediatr. Otorhinolaryngol. 2001, 58, 215–221. [Google Scholar] [CrossRef]
- Alkhunaizi, E.; Brosh, R.M., Jr.; Alkuraya, F.S.; Chitayat, D. Warsaw Syndrome. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. [Google Scholar]
- Rabin, R.; Hirsch, Y.; Johansson, M.M.; Ekstein, J.; Zeevi, D.A.; Keena, B.; Zackai, E.H.; Pappas, J. Study of carrier frequency of Warsaw breakage syndrome in the Ashkenazi Jewish population and presentation of two cases. Am. J. Med. Genet. Part A 2019, 179, 2144–2151. [Google Scholar] [CrossRef]
- Alkhunaizi, E.; Shaheen, R.; Bharti, S.K.; Joseph-George, A.M.; Chong, K.; Abdel-Salam, G.M.H.; Alowain, M.; Blaser, S.I.; Papsin, B.C.; Butt, M.; et al. Warsaw breakage syndrome: Further clinical and genetic delineation. Am. J. Med. Genet. Part A 2018, 176, 2404–2418. [Google Scholar] [CrossRef]
- Bottega, R.; Napolitano, L.M.R.; Carbone, A.; Cappelli, E.; Corsolini, F.; Onesti, S.; Savoia, A.; Gasparini, P.; Faletra, F. Two further patients with Warsaw breakage syndrome. Is a mild phenotype possible? Mol. Genet. Genom. Med. 2019, 7, e639. [Google Scholar] [CrossRef]
- Brotto, D.; Manara, R.; Scimemi, P.; Sorrentino, F.; Montino, S.; Maritan, F.; Caserta, E.; Lovo, E.; Martini, A.; Santarelli, R.; et al. An audiological perspective on “Two further patients with Warsaw breakage syndrome. Is a mild phenotype possible?”. Mol. Genet. Genom. Med. 2020, 8. [Google Scholar] [CrossRef]
- Talenti, G.; Robson, C.; Severino, M.S.; Alves, C.A.; Chitayat, D.; Dahmoush, H.; Smith, L.; Muntoni, F.; Blaser, S.I.; D’Arco, F. Characteristic Cochlear Hypoplasia in Patients with Walker-Warburg Syndrome: A Radiologic Study of the Inner Ear in α-Dystroglycan-Related Muscular Disorders. Am. J. Neuroradiol. 2021, 42, 167–172. [Google Scholar] [CrossRef]
- Hurd, E.A.; Poucher, H.K.; Cheng, K.; Raphael, Y.; Martin, D.M. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development 2010, 137, 3139–3150. [Google Scholar] [CrossRef] [Green Version]
- Brotto, D.; Manara, R.; Gallo, S.; Sorrentino, F.; Bovo, R.; Trevisi, P.; Martini, A. Comments on “hearing restoration in cochlear nerve deficiency: The choice between cochlear implant or auditory brainstem implant, a meta-analysis”. Otol. Neurotol. 2019, 40, 543–544. [Google Scholar] [CrossRef]
- Kaiser, M.; Wojahn, I.; Rudat, C.; Lüdtke, T.H.; Christoffels, V.M.; Moon, A.; Kispert, A.; Trowe, M.O. Regulation of otocyst patterning by Tbx2 and Tbx3 is required for inner ear morphogenesis in the mouse. Development 2021, 148. [Google Scholar] [CrossRef]
- Gavalas, A.; Studer, M.; Lumsden, A.; Rijli, F.M.; Krumlauf, R.; Chambon, P. Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development 1998, 125, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.; Goold, A.; Egelhoff, J.; Benton, C. Partial absence of the posterior semicircular canal in Alagille syndrome: CT findings. Pediatr. Radiol. 2006, 36, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Guyot, J.P.; Gacek, R.R.; DiRaddo, P. The temporal bone anomaly in CHARGE association. Arch. Otolaryngol. Head Neck Surg. 1987, 113, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Sando, I.; Bergstrom, L.; Wood, R.P., 2nd; Hemenway, W.G. Temporal bone findings in trisomy 18 syndrome. Arch. Otolaryngol. Head Neck Surg. 1970, 91, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Higashi, K.; Matsuki, C.; Sarashina, N. Aplasia of posterior semicircular canal in Waardenburg syndrome type II. J. Otolaryngol. 1992, 21, 262–264. [Google Scholar]
- Okuno, T.; Takahashi, H.; Shibahara, Y.; Hashida, Y.; Sando, I. Temporal bone histopathologic findings in Alagille’s syndrome. Arch. Otolaryngol. Head Neck Surg. 1990, 116, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Alagille, D.; Odièvre, M.; Gautier, M.; Dommergues, J.P. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J. Pediatr. 1975, 86, 63–71. [Google Scholar] [CrossRef]
- Li, L.; Krantz, I.D.; Deng, Y.; Genin, A.; Banta, A.B.; Collins, C.C.; Qi, M.; Trask, B.J.; Kuo, W.L.; Cochran, J.; et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 1997, 16, 243–251. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sennaroglu (2002) | Sennaroglu (2017) | Subgroups (2017) | Description |
|---|---|---|---|
| Michel deformity | Complete labyrinthine aplasia | With hypoplastic or aplastic petrous bone | Absence of the whole cochleovestibular structure |
| With otic capsule | |||
| Without otic capsule | |||
| Rudimentary otocyst | No subgroups | Incomplete and small representation of the otocyst without an internal auditory canal | |
| Cochlear aplasia | Cochlear aplasia | With normal labyrinth | Absence of the cochlea |
| With a dilated vestibule (CAVD) | Absence of the cochlea, bust the vestibule is enlarged | ||
| Common cavity deformity | Common cavity | No subgroups | Single, round chamber, representing cochlea and vestibule without any differentiation between them |
| Incomplete partition type 1 | Incomplete partitions of the cochlea (differentiation of cochlea and vestibule, normal external dimensions) | Incomplete partition type 1 | The entire modiolus and interscalar septa are absent, resulting in a cystic appearance of the cochlea |
| Incomplete partition type 2 | Incomplete partition type 2 | Absence of the apical part of the modiolus and corresponding interscalar septa, fusion of middle and apical turns | |
| Incomplete partition type 3 | Absent modiolus but complete interscalar septa, enlarged internal acoustic canal | ||
| Cochlear hypoplasia (corresponding to CH-I) | Cochlear hypoplasia (external cochlear dimensions are smaller than normal) | Type1 | Cochlea is like a small bud |
| Type 2 | Defective modiolus and interscalar septa, normal external outline | ||
| Type 3 | Short modiolus and less than 2 turns, reduced length of the interscalar septa | ||
| Type 4 | Normal basal turn, middle and apical turns are severely hypoplastic and located anteriorly and medially rather than in the center |
| Type of Malformation | Possible Related Syndrome | Genes Involved |
|---|---|---|
| Complete labyrinthine aplasia | Wildervanck syndrome | - |
| LAMM syndrome | FGF3 | |
| HOXA1 mutation syndrome | HOXA1 | |
| Rudimentary otocyst | - | - |
| Common cavity | Bosley–Salih–Alorainy syndrome | HOXA1 |
| 22q11 deletion syndrome | - | |
| - | ROR1 | |
| Cochlear aplasia | - | PAX2 (animal studies) |
| Incomplete partition type 1 | - | - |
| Incomplete partition type 2 | Pendred syndrome | SLC26A4 |
| - | FOXI1 | |
| - | KCNJ10 | |
| Incomplete partition type 3 | - | POUF3F4 |
| Cochlear hypoplasia | Branchio-oto-renal syndrome | - |
| Oculo-auriculo-vertebral spectrum disorder | - | |
| Warsaw breakage syndrome | DDX11 | |
| Pallister–Hall syndrome | - | |
| - | SIX1 | |
| - | CHD7 (animal studies) | |
| - | TBX2 (animal studies) | |
| - | HOXA1/HOXB1 (animal studies) | |
| Cochlear hypoplasia Type 1 | - | - |
| Cochlear hypoplasia Type 2 | - | - |
| Cochlear hypoplasia Type 3 | Waardenburg syndrome | SOX10 |
| Cochlear hypoplasia Type 4 | ⲁ-Dystroglycan-related muscular disorders (Walker–Warburg syndrome) | - |
| Posterior labyrinth anomalies | CHARGE syndrome | - |
| Trisomy 13 | - | |
| Trisomy 18 | - | |
| Waardenburg syndrome type II | - | |
| Alagille syndrome | JAG1 | |
| Warsaw breakage syndrome | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brotto, D.; Sorrentino, F.; Cenedese, R.; Avato, I.; Bovo, R.; Trevisi, P.; Manara, R. Genetics of Inner Ear Malformations: A Review. Audiol. Res. 2021, 11, 524-536. https://doi.org/10.3390/audiolres11040047
Brotto D, Sorrentino F, Cenedese R, Avato I, Bovo R, Trevisi P, Manara R. Genetics of Inner Ear Malformations: A Review. Audiology Research. 2021; 11(4):524-536. https://doi.org/10.3390/audiolres11040047
Chicago/Turabian StyleBrotto, Davide, Flavia Sorrentino, Roberta Cenedese, Irene Avato, Roberto Bovo, Patrizia Trevisi, and Renzo Manara. 2021. "Genetics of Inner Ear Malformations: A Review" Audiology Research 11, no. 4: 524-536. https://doi.org/10.3390/audiolres11040047
APA StyleBrotto, D., Sorrentino, F., Cenedese, R., Avato, I., Bovo, R., Trevisi, P., & Manara, R. (2021). Genetics of Inner Ear Malformations: A Review. Audiology Research, 11(4), 524-536. https://doi.org/10.3390/audiolres11040047