
Highlights on the Luspatercept Treatment in Thalassemia


Abstract
:1. Introduction
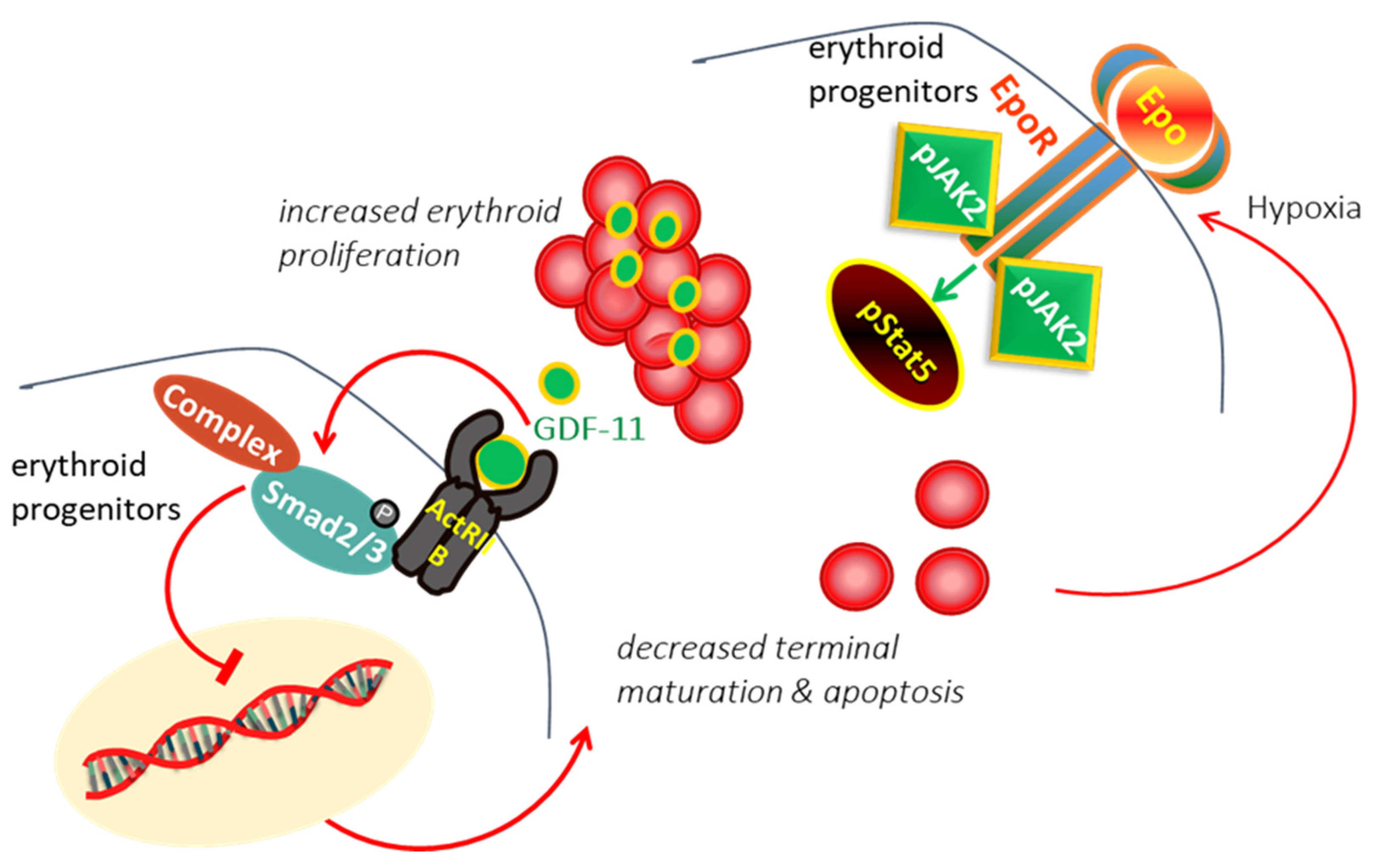
2. Ineffective Erythropoiesis in β-Thalassemia
3. Modulating Erythroid Differentiation by ActRII Ligand Traps
4. Pharmacokinetics of Luspatercept
5. Clinical Studies with Luspatercept
6. Current Status, Challenges, and Future Directions of Luspatercept
7. Conclusions
Funding
Conflicts of Interest
References
- Kattamis, A.; Kwiatkowski, J.L.; Aydinok, Y. Thalassaemia. Lancet 2022, 399, 2310–2324. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Daar, S.; Taher, A.T. Morbidity-free survival and hemoglobin level in non-transfusion-dependent β-thalassemia: A 10-year cohort study. Ann. Hematol. 2022, 101, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Kattamis, A.; Viprakasit, V.; Sutcharitchan, P.; Pariseau, J.; Laadem, A.; Jessent-Ciaravino, V.; Taher, A. Quality of life in patients with β-thalassemia: A prospective study of transfusion-dependent and non-transfusion-dependent patients in Greece, Italy, Lebanon, and Thailand. Am. J. Hematol. 2019, 94, E261–E264. [Google Scholar] [CrossRef] [Green Version]
- Cappellini, M.D.; Porter, J.B.; Viprakasit, V.; Taher, A.T. A paradigm shift on beta-thalassaemia treatment: How will we manage this old disease with new therapies? Blood Rev. 2018, 32, 300–311. [Google Scholar] [CrossRef]
- A Phase 2 Study to Evaluate the Safety and Tolerability of IMR-687 in Subjects with Beta Thalassemia. Available online: https://ir.imaratx.com/news-releases/news-release-details/imara-announces-results-interim-analyses-tovinontrine-imr-687 (accessed on 5 April 2022).
- Taher, A.T.; Karakas, Z.; Cassinerio, E.; Siritanaratkul, N.; Kattamis, A.; Maggio, A.; Rivella, S.; Hollaender, N.; Mahuzier, B.; Gadbaw, B.; et al. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: Results from a phase 2a study. Blood 2018, 131, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Gardenghi, S.; Grady, R.W.; Rivella, S. Anemia, ineffective erythropoiesis, and hepcidin: Interacting factors in abnormal iron metabolism leading to iron overload in beta-thalassemia. Hematol. Oncol. Clin. N. Am. 2010, 24, 1089–1107. [Google Scholar] [CrossRef] [Green Version]
- Lal, A.; Voskaridou, E.; Flevari, P.; Tahe, A.; Chew, L.; Valone, F.; Gupta, S.; Viprakasit, V. A hepcidin mimetic, PTG-300, demonstrates pharmacodynamic effects indicating reduced iron availability in transfusion dependent beta-thalassemia subjects. Hemasphere 2020, 4, 298. [Google Scholar]
- Taher, A.; Kourakli-Symeonidis, A.; Tantiworawit, A.; Wong, P.; Szecsödy, P. Safety and preliminary pharmacodynamics effects of the ferroportin inhibitor vamifeport (VIT-2763) in patients with non-transfusion dependent beta-thalassemia (NTDT): Results from a phase 2a study. HemaSphere 2022, 6, 350. [Google Scholar]
- Vadolas, J.; Ng, G.Z.; Kysenius, K.; Crouch, P.J.; Dames, S.; Eisermann, M.; Nualkaew, T.; Vilcassim, S.; Schaeper, U.; Grigoriadis, G. SLN124, a GalNac-siRNA targeting transmembrane serine protease 6, in combination with deferiprone therapy reduces ineffective erythropoiesis and hepatic iron-overload in a mouse model of β-thalassaemia. Br. J. Haematol. 2021, 194, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.H.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E.P. Results from a phase 2, open label, multicenter study of the oral pyruvate kinase activator mitapivat in adults with non-transfusion dependent alpha- or beta-thalassemia. Hemasphere 2021, 5 (Suppl. 2), 92. [Google Scholar]
- Centis, F.; Tabellini, L.; Lucarelli, G.; Buffi, O.; Tonucci, P.; Persini, B.; Annibali, M.; Emiliani, R.; Iliescu, A.; Rapa, S.; et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with β-thalassemia major. Blood 2000, 96, 3624–3629. [Google Scholar] [CrossRef]
- Mathias, L.A.; Fisher, T.C.; Zeng, L.; Meiselman, H.J.; Weinberg, K.I.; Hiti, A.L.; Malik, P. Ineffective erythropoiesis in β-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp. Hematol. 2000, 28, 1343–1353. [Google Scholar] [CrossRef]
- Libani, I.V.; Guy, E.C.; Melchiori, L.; Schiro, R.; Ramos, P.; Breda, L.; Scholzen, T.; Chadburn, A.; Liu, Y.; Kernbach, M.; et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in β-thalassemia. Blood 2008, 112, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikonomidou, P.R.; Rivella, S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018, 32, 130–143. [Google Scholar] [CrossRef]
- Wojchowski, D.M.; Gregory, R.C.; Miller, C.P.; Pandit, A.K.; Pircher, T.J. Signal transduction in the erythropoietin receptor system. Exp. Cell Res. 1999, 253, 143–156. [Google Scholar] [CrossRef]
- Rivella, S.; Melchiori, L.; Gardenghi, S. β-thalassemia: HiJAKing ineffective erythropoiesis and iron overload. Adv. Hematol. 2010, 2010, 938640. [Google Scholar] [CrossRef] [Green Version]
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in β-thalassemia. Nat. Med. 2014, 20, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.V.S.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef]
- Söderberg, S.S.; Karlsson, G.; Karlsson, S. Complex and context dependent regulation of hematopoiesis by tgf-β superfamily signaling. Ann. N. Y. Acad. Sci. 2009, 1176, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.A.; Li, R.; Ramanathan, H.N.; Bhasin, M.; Pearsall, R.S.; Kumar, R.; Suragani, R.N.V.S. Smad2/3-pathway ligand trap luspatercept enhances erythroid differentiation in murine β-thalassaemia by increasing GATA-1 availability. J. Cell Mol. Med. 2020, 24, 6162–6177. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.V.S.; Cawley, S.M.; Li, R.; Wallner, S.; Alexander, M.J.; Mulivor, A.W.; Gardenghi, S.; Rivella, S.; Grinberg A., V.; Pearsall, R.S.; et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β-thalassemia. Blood 2014, 123, 3864–3872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Kassir, N.; Laadem, A.; Giuseppi, A.C.; Shetty, J.; Maxwell, S.E.; Sriraman, P.; Ritland, S.; Linde, P.G.; Budda, B.; et al. Population Pharmacokinetics and Exposure-Response Relationship of Luspatercept, an Erythroid Maturation Agent, in Anemic Patients With β-Thalassemia. J. Clin. Pharmacol. 2021, 61, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Attie, K.M.; Allison, M.J.; Mcclure, T.; Boyd, I.E.; Wilson, D.M.; Pearsall, A.E.; Sherman, M.L. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am. J. Hematol. 2014, 89, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reblozyl. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/reblozyl-epar-product-information_en.pdf (accessed on 6 January 2023).
- Piga, A.; Perrotta, S.; Gamberini, M.R.; Voskaridou, E.; Melpignano, A.; Filosa, A.; Caruso, V.; Pietrangelo, A.; Longo, F.; Tartaglione, I.; et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with b-thalassemia. Blood 2019, 133, 1279–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappellini, M.D.; Porter, J.; Origa, R.; Forni, G.L.; Voskaridou, E.; Galactéros, F.; Taher, A.T.; Arlet, J.B.; Ribeil, J.A.; Garbowski, M.; et al. Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: A phase II, open-label, dose-finding study. Haematologica 2019, 104, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.-K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Cappellini, M.D.D.; Taher, A.T.; Porter, J.B.; Kuo, K.H.M.; Coates, T.D.; Voskaridou, E.; Forni, G.L.; Perrotta, S.; Khelif, A. Longer-term analysis of the efficacy of luspatercept versus placebo in patients with transfusion-dependent beta-thalassemia enrolled in the Believe study. HemaSphere 2022, 6, 270. [Google Scholar] [CrossRef]
- Hermine, O.; Cappellini, M.D.D.; Taher, A.T.; Coates, T.D.; Viprakasit, V.; Kattamis, A.; Shetty, J.K.; Weisskopf, M.B.; Holot, N.; Vodala, S.; et al. Effect of Luspatercept on Red Blood Cell (RBC) Transfusion Burden, Iron Chelation Therapy (ICT), and Iron Overload in Adults with Transfusion-Dependent β-Thalassemia (TDT) from the BELIEVE Trial: A Long-Term Analysis. Blood 2022, 140, 8215–8217. [Google Scholar] [CrossRef]
- Sheth, S.; Hermine, O.; Taher, A.T.; Kuo, K.H.M.; Porter, J.B.; Piga, A.; Coates, T.D.; Kattamis, A.; Medlin, L.F.; Kuo, W.-L.; et al. Effect of Luspatercept in β-Thalassemia Patients with β0/β0 Genotype: A Subgroup Analysis of the BELIEVE Study. Blood 2022, 140, 1946–1948. [Google Scholar] [CrossRef]
- Viprakasit, V.; Cappellini, M.D.; Porter, J.B.; Kuo, K.H.M.; Coates, T.D.; Voskaridou, E.; Pinto, V.M.; Tartaglione, I.; Khelif, A.; Lal, A.; et al. P1518: Long-Term Safety Results of the Believe Study of Luspatercept in Adults With Βeta-Thalassemia. HemaSphere 2022, 6, 1399–1400. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D.; Kattamis, A.; Voskaridou, E.; Perrotta, S.; Piga, A.G.; Filosa, A.; Porter, J.B.; Coates, T.D.; Forni, G.L.; et al. Luspatercept for the treatment of anaemia in non-transfusion-dependent β-thalassaemia (BEYOND): A phase 2, randomised, double-blind, multicentre, placebo-controlled trial. Lancet Haematol. 2022, 9, e733–e744. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Coates, T.D.; Musallam, K.M.; Buerki, J.V.; Patturajan, M.; Holot, N.; Aydinok, A. A Phase 2a Study Evaluating the Safety and Pharmacokinetics (PK) of Luspatercept in Pediatric Patients with Transfusion-Dependent β-Thalassemia (TDT). Blood 2021, 138, 4161. [Google Scholar] [CrossRef]


{kind=link}
| Protocol no | Phase | Planned/Treated | Dose (mg/kg) | Duration | Erythroid Response |
|---|---|---|---|---|---|
| NCT01432717 | 1 | 32 PM women | 0.0625–0.25 | 1 month (Q2W) | Hb ≥ 1 g/dL, 83% (0.25 mg/kg) |
| NCT01749540 | 2 | 64/64 (NTDT, TDT, ≥18y) | 0.2–1.25 | 15 weeks (Q3W) | ∫ NTDT; 58% achieved Hb ≥ 1.5 g/dL for 2 weeks, 71% Hb ≥ 1.0 g/dL for 12 weeks. ∫ TDT; transfusion reduction ≥ 20, 33, and 50% at 81, 72, and 63%, respectively, for 12 weeks |
| NCT02268409 | 2 | 64/51 (NTDT, TDT, ≥18y) | 0.8–1.25 | 60 months (Q3W) | |
| NCT02604433 [BELIEVE] | 3 | 336/332 (TDT, ≥18y, luspatercept vs. placebo 224:112) open-label LTFU ongoing | 1.0–1.25 | 48 weeks LTFU:60 months (Q3W) | ≥33% reduction: weeks 13–24; 21.4% in luspatercept vs. 4.5% placebo ≥50% reduction: weeks 13–24; 7.6% in luspatercept vs. 1.8% placebo and ≥50% reduction: weeks 37–48; 10.3% in luspatercept vs. 0.9% placebo ≥33% reduction: any 24 weeks; 41.1% in luspatercept vs. 2.7% placebo |
| NCT03342404 [BEYOND] | 2 | 150/145 (NTDT, ≥18y, luspatercept vs. placebo 96:49) | 1.0–1.25 | 48 weeks (Q3W) | Hb ≥ 1.0 g/dL: weeks 13–24; 77% in luspatercept vs. 0% placebo (p < 0.0001) Mean Hb change (g/dL) from baseline weeks 13–24; 1.48 in luspatercept vs. 0.07 placebo (p < 0.0001) Hb ≥ 1.0 g/dL: weeks 37–48; 71% in luspatercept vs. 2% placebo (p < 0.0001) |
| NCT04143724 | 2 | 54/- (TDT, 6– < 18y) | 0.75–1.25 | 1 year LTFU: 5 year | Started patients’ recruitment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aydinok, Y. Highlights on the Luspatercept Treatment in Thalassemia. Thalass. Rep. 2023, 13, 77-84. https://doi.org/10.3390/thalassrep13010008
Aydinok Y. Highlights on the Luspatercept Treatment in Thalassemia. Thalassemia Reports. 2023; 13(1):77-84. https://doi.org/10.3390/thalassrep13010008
Chicago/Turabian StyleAydinok, Yesim. 2023. "Highlights on the Luspatercept Treatment in Thalassemia" Thalassemia Reports 13, no. 1: 77-84. https://doi.org/10.3390/thalassrep13010008
APA StyleAydinok, Y. (2023). Highlights on the Luspatercept Treatment in Thalassemia. Thalassemia Reports, 13(1), 77-84. https://doi.org/10.3390/thalassrep13010008