
Dysregulation of Iron Homeostasis in β-Thalassemia and Impaired Neutrophil Activity

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Hematological and Biochemical Parameters
2.3. Neutrophil Purification
2.4. RNA Extraction and Real-Time qPCR
2.5. Oxidative Burst
2.6. Phagocytosis Activity
2.7. Statistical Analysis
3. Results
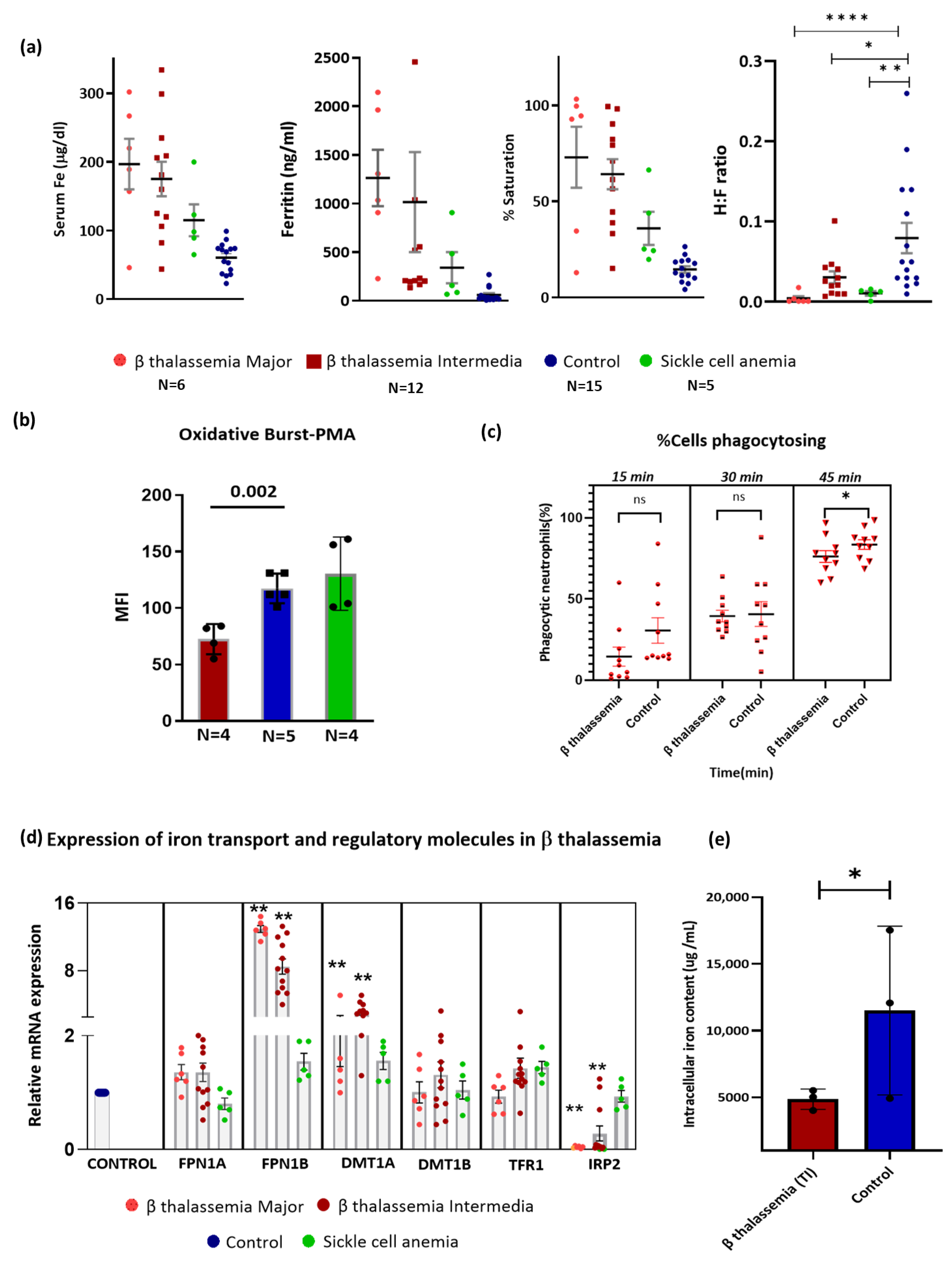
3.1. Hematological and Biochemical Parameters in the Study Participants
3.2. Neutrophils of β-Thalassemia Patients Exhibited Reduced Burst Activity and Phagocytosis
3.3. Iron Metabolism in Neutrophils of β-Thalassemia
3.4. Association of Systemic Iron Status with Neutrophil Function and Neutrophil Iron Metabolism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biswas, S.; Smrity, S.Z.; Bhuia, M.S.; Sonia, F.A.; Aktar, M.A.; Chowdhury, R.; Islam, T.; Islam, M.T.; Gonçalves Alencar, G.; Paulo, C.L.R.; et al. Beta-Thalassemia: A Pharmacological Drug-Based Treatment. Drugs Drug Candidates 2024, 3, 126–147. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Cappellini, M.D.; De Stefano, P.; Del Vecchio, G.C.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; Origa, R.; Piga, A.; Romeo, M.A.; et al. Survival and Complications in Thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 40–47. [Google Scholar] [CrossRef]
- Namazi Bayegi, S.; Ali Hamidieh, A.; Behfar, M.; Saghazadeh, A.; Bozorgmehr, M.; Tajik, N.; Delbandi, A.-A.; Delavari, S.; Shekarabi, M.; Rezaei, N. Unbalanced T-cell subsets in pediatric patients with beta-thalassemia. Hum. Immunol. 2023, 84, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Vento, S.; Cainelli, F.; Cesario, F. Infections and thalassaemia. Lancet Infect. Dis. 2006, 6, 226–233. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun. Signal. 2019, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Sae-Khow, K.; Charoensappakit, A.; Leelahavanichkul, A. Neutrophil Diversity (Immature, Aged, and Low-Density Neutrophils) and Functional Plasticity: Possible Impacts of Iron Overload in β-Thalassemia. Int. J. Mol. Sci. 2024, 25, 10651. [Google Scholar] [CrossRef]
- Thiengtavor, C.; Siriworadetkun, S.; Paiboonsukwong, K.; Fucharoen, S.; Pattanapanyasat, K.; Vadolas, J.; Svasti, S.; Chaichompoo, P. Increased ferritin levels in non-transfusion-dependent β°-thalassaemia/HbE are associated with reduced CXCR2 expression and neutrophil migration. Br. J. Haematol. 2020, 189, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Cantinieaux, B.; Hariga, C.; Ferster, A.; De Maertelaere, E.; Toppet, M.; Fondu, P. Neutrophil dysfunctions in thalassaemia major: The role of cell iron overload. Eur. J. Haematol. 1987, 39, 28–34. [Google Scholar] [CrossRef]
- van Asbeck, B.S.; Marx, J.J.; Struyvenberg, A.; van Kats, J.H.; Verhoef, J. Effect of iron (III) in the presence of various ligands on the phagocytic and metabolic activity of human polymorphonuclear leukocytes. J. Immunol. 1984, 132, 851–856. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Skoutelis, A.T.; Lianou, E.; Papavassiliou, T.; Karamerou, A.; Politi, K.; Bassaris, H.P. Defective phagocytic and bactericidal function of polymorphonuclear leucocytes in patients with β-thalassaemia major. J. Infect. 1984, 8, 118–122. [Google Scholar] [CrossRef]
- Cantinieaux, B.; Hariga, C.; Fondu, P.; Ferster, A.; Toppet, M. Desferrioxamine improves neutrophil phagocytosis in thalassemia major. Am. J. Hematol. 1990, 35, 13–17. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed]
- Mangla, A.; Ehsan, M.; Agarwal, N.; Maruvada, S. Sickle Cell Anemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK482164/ (accessed on 13 November 2024).
- de Azevedo, J.T.C.; Malmegrim, K.C.R. Immune mechanisms involved in sickle cell disease pathogenesis: Current knowledge and perspectives. Immunol. Lett. 2020, 224, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Akinbo, D.B.; Ajayi, O.I.; Eluji, O.M.; Olatunji, I.; Okoroloko, T.M. Impaired phagocytosis and oxidative respiratory burst activity in sickle cell anemia leukocytes. J. Taibah Univ. Med. Sci. 2024, 19, 867. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Ekwattanakit, S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol./Oncol. Clin. N. Am. 2018, 32, 193–211. [Google Scholar] [CrossRef]
- Singh, P.; Shaikh, S.; Parmar, S.; Gupta, R. Current Status of β-Thalassemic Burden in India. Hemoglobin 2023, 47, 181–190. [Google Scholar] [CrossRef]
- Rao, P.; Raj, E.A.; Natesan, S.; Gudi, N. Prevalence of Sickle cell disease, Sickle cell trait and HBS-beta-thalassemia in India: A systematic review and Meta-analysis. Clin. Epidemiol. Glob. Health 2024, 28, 101678. [Google Scholar] [CrossRef]
- Chuncharunee, S.; Teawtrakul, N.; Siritanaratkul, N.; Chueamuangphan, N. Review of disease-related complications and management in adult patients with thalassemia: A multi-center study in Thailand. PLoS ONE 2019, 14, e0214148. [Google Scholar] [CrossRef]
- Renassia, C.; Louis, S.; Cuvellier, S.; Boussetta, N.; Deschemin, J.-C.; Borderie, D.; Bailly, K.; Poupon, J.; Dang, P.M.-C.; El-Benna, J.; et al. Neutrophils from hereditary hemochromatosis patients are protected from iron excess and are primed. Blood Adv. 2020, 4, 3853–3863. [Google Scholar] [CrossRef]
- Siwaponanan, P.; Siegers, J.Y.; Ghazali, R.; Ng, T.; McColl, B.; Ng, G.Z.-W.; Sutton, P.; Wang, N.; Ooi, I.; Thiengtavor, C.; et al. Reduced PU.1 expression underlies aberrant neutrophil maturation and function in β-thalassemia mice and patients. Blood 2017, 129, 3087–3099. [Google Scholar] [CrossRef] [PubMed]
- Buttari, B.; Profumo, E.; Caprari, P.; Massimi, S.; Sorrentino, F.; Maffei, L.; Gabbianelli, M.; Riganò, R. Phenotypical and functional abnormalities of circulating neutrophils in patients with β-thalassemia. Ann. Hematol. 2020, 99, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Kurtoglu, E.; Ugur, A.; Baltaci, A.K.; Mogolkoc, R.; Undar, L. Activity of neutrophil NADPH oxidase in iron-deficient anemia. Biol. Trace Elem. Res. 2003, 96, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cohort | Age (Years) | Gender | Hb (g/dL) | MCV (fL) | Neutrophil Count (cells/µL) | Ferritin (ng/mL) | Hepcidin (ng/mL) | sTfR (mg/dL) |
|---|---|---|---|---|---|---|---|---|
| TM (N = 6) | 16 ± 7.8 | M:1 F:5 | 8.3 ± 2.1 * | 84.1 ± 3.3 | 3.37 × 103 ± 2.11 | 1170.2 (229–2144.6) | 18.1 (1–48.4) | 16.7 (10.5–34.4) |
| TI (N = 12) | 24.5 ± 9.5 | M:8 F:4 | 8 ± 2.1 * | 75 ± 13.2 | 4.52 × 103 ± 1.5 | 220 (167–2457.4) | 3.7 (1–20) | 16 (5.24–30) |
| SCA (N = 5) | 19.6 ± 5 | M:2 F:3 | 10.1 ± 1.1 | 75.6 ± 5.1 | 5.58 × 103 ± 2.4 | 157.2 (68.3–2907.4) | 2.1 (1–16.14) | 12 (8.5–13.15) |
| Control (N = 15) | 28.5 ± 5 | M:6 F:9 | 13.2 ± 1.8 | 82.6 ± 6.1 | 5.48 × 103 ± 1.3 | 29.9 (16–269.4) | 1.66 (1–8.2) | 4.2 (2.3–7.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santhakumar, S.; Stephen, L.; Barade, A.; Kulkarni, U.; George, B.; Edison, E.S. Dysregulation of Iron Homeostasis in β-Thalassemia and Impaired Neutrophil Activity. Thalass. Rep. 2025, 15, 4. https://doi.org/10.3390/thalassrep15020004
Santhakumar S, Stephen L, Barade A, Kulkarni U, George B, Edison ES. Dysregulation of Iron Homeostasis in β-Thalassemia and Impaired Neutrophil Activity. Thalassemia Reports. 2025; 15(2):4. https://doi.org/10.3390/thalassrep15020004
Chicago/Turabian StyleSanthakumar, Sreenithi, Leo Stephen, Aruna Barade, Uday Kulkarni, Biju George, and Eunice S. Edison. 2025. "Dysregulation of Iron Homeostasis in β-Thalassemia and Impaired Neutrophil Activity" Thalassemia Reports 15, no. 2: 4. https://doi.org/10.3390/thalassrep15020004
APA StyleSanthakumar, S., Stephen, L., Barade, A., Kulkarni, U., George, B., & Edison, E. S. (2025). Dysregulation of Iron Homeostasis in β-Thalassemia and Impaired Neutrophil Activity. Thalassemia Reports, 15(2), 4. https://doi.org/10.3390/thalassrep15020004