
Unilateral Retinitis Pigmentosa Associated with Possible Ciliopathy and a Novel Mutation

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
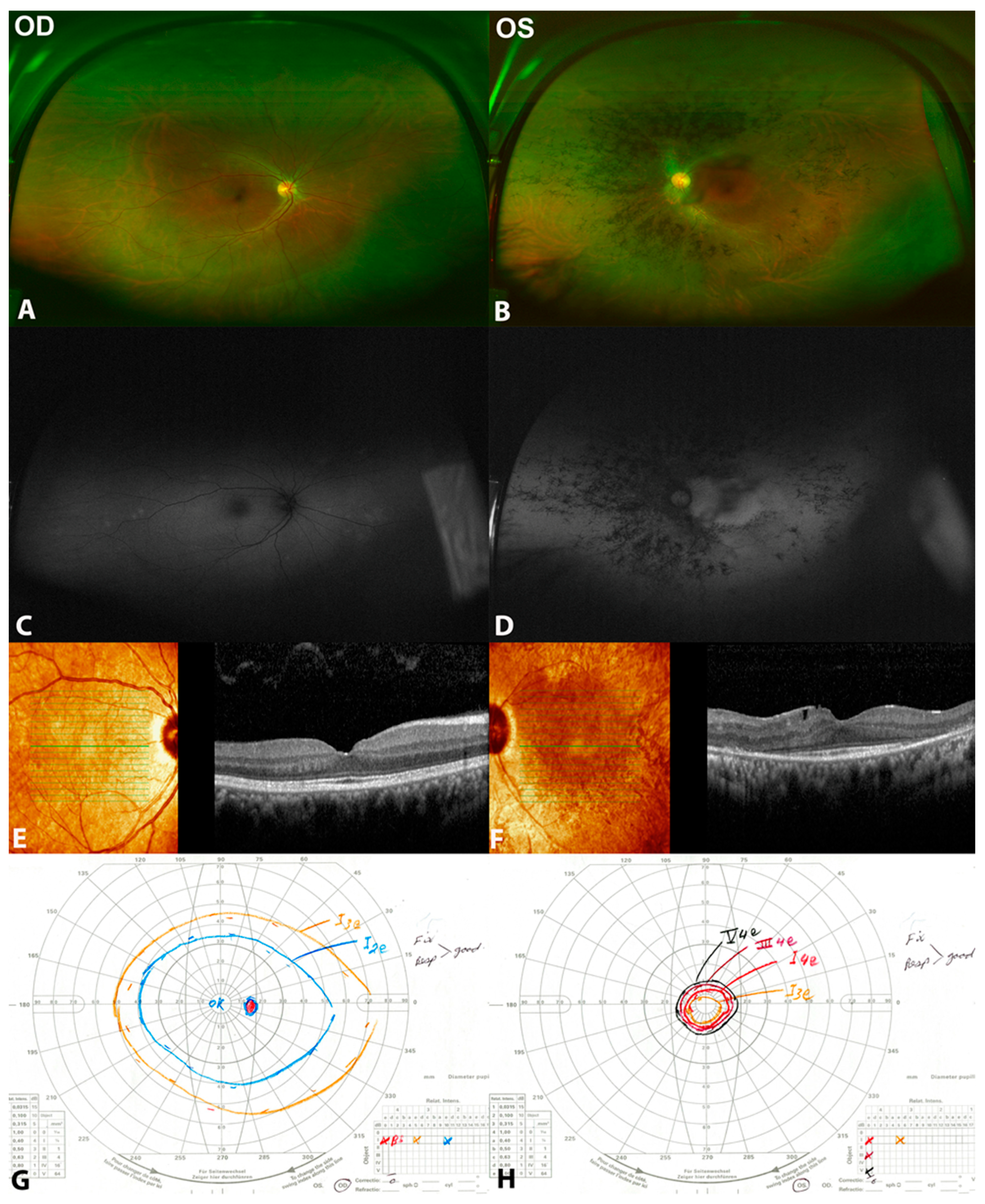
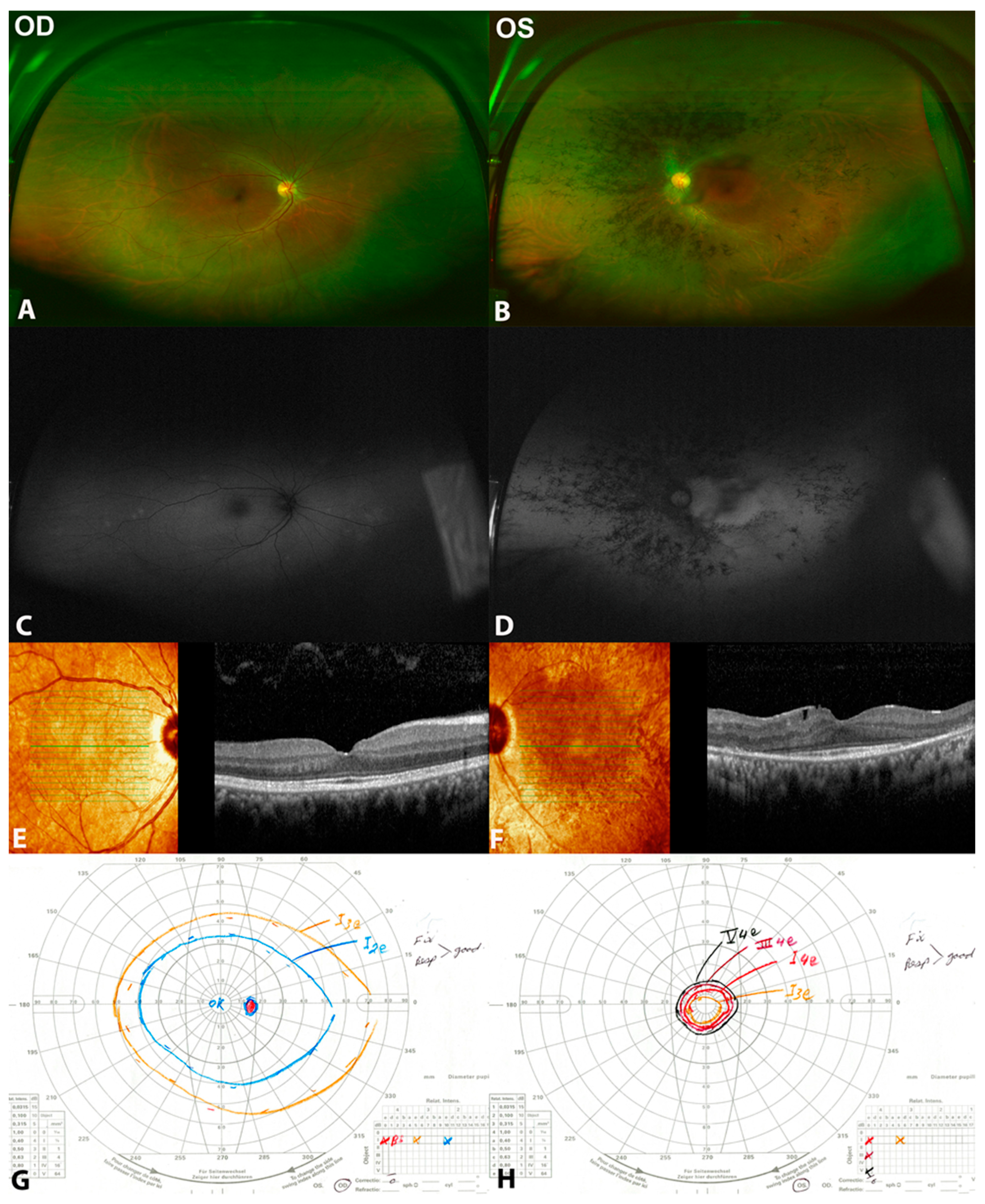
3.1. Patient 1
3.2. Patient 2
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bin, P.; Haifeng, M.; Yiqiao, X. Unilateral retinitis pigmentosa. J. Anim. Vet. Adv. 2013, 12, 779–781. [Google Scholar] [CrossRef]
- Nazar, C.; Feldman, M.; González, R.; Espinoza, R. Unilateral retinitis pigmentosa. A case report. Arch. Soc. Española Oftalmol. 2017, 92, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.F. Unilateral retinitis pigmentosa and cone-rod dystrophy. Clin. Ophthalmol. 2009, 3, 263–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Errera, M.-H.; Robson, A.G.; Wong, T.; Hykin, P.G.; Pal, B.; Sagoo, M.S.; Pavesio, C.E.; Moore, A.T.; Webster, A.R.; MacLaren, R.E.; et al. Unilateral pigmentary retinopathy: A retrospective case series. Acta Ophthalmol. 2019, 97, e601–e617. [Google Scholar] [CrossRef]
- Marsiglia, M.; Duncker, T.; Peiretti, E.; Brodie, S.E.; Tsang, S.H. Unilateral retinitis pigmentosa: A proposal of genetic pathogenic mechanisms. Eur. J. Ophthalmol. 2012, 22, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, R.; Holder, G.E.; Moore, A.T.; Webster, A.R. Unilateral retinitis pigmentosa occurring in an individual with a germline mutation in the RP1 gene. Arch Ophthalmol. 2011, 129, 954–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, P.Y.; Jeganathan, V.S.E.; Wright, A.F.; Cackett, P. Unilateral retinitis pigmentosa occurring in an individual with a mutation in the CLRN1 gene. BMJ Case Rep. 2018, 2018, 1–5. [Google Scholar] [CrossRef]
- Takahashi, V.K.L.; Xu, C.L.; Takiuti, J.T.; Apatoff, M.B.L.; Duong, J.K.; Mahajan, V.B.; Tsang, S.H. Comparison of structural progression between ciliopathy and non-ciliopathy associated with autosomal recessive retinitis pigmentosa. Orphanet J. Rare Dis. 2019, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Magliyah, M.; Dhibi, H.A.; Schatz, P. Analysis of retinal function and structure in autosomal recessive retinal-renal ciliopathy. Eye 2020, 34, 2135–2137. [Google Scholar] [CrossRef]
- Racette, L.; Fischer, M.; Bebie, H.; Holló, G.; Johnson, C.A.; Matsumoto, C. Visual Field Digest. A Guide to Perimetry and the Octopus Perimeter; Haag-Streit AG: Köniz, Switzerland, 2016; p. 291. [Google Scholar]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef]
- Magliyah, M.S.; Alsulaiman, M.; Schatz, P.; Nowilaty, R. Evolution of macular hole in enhanced S-cone syndrome. Doc. Ophthalmol. 2021, 142, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, B.; Lerolle, N.; Radermacher, P.; Asfar, P. Target blood pressure in sepsis: Between a rock and a hard place. Crit. Care 2013, 17, 126. [Google Scholar] [CrossRef] [Green Version]
- Bawankar, P.; Deka, H.; Barman, M.; Bhattacharjee, H.; Soibam, R. Unilateral retinitis pigmentosa: Clinical and electrophysiological diagnosis. Can. J. Ophthalmol. 2018, 53, e94–e97. [Google Scholar] [CrossRef]
- Patel, N.; Aldahmesh, M.A.; Alkuraya, H.; Anazi, S.; Alsharif, H.; Khan, A.O.; Sunker, A.; Al-Mohsen, S.; Abboud, E.B.; Nowilaty, S.R.; et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 2016, 18, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branham, K.; Matsui, H.; Biswas, P.; Guru, A.A.; Hicks, M.; Suk, J.J.; Li, H.; Jakubosky, D.; Long, T.; Telenti, A.; et al. Establishing the involvement of the novel gene AGBL5 in retinitis pigmentosa by whole genome sequencing. Physiol. Genom. 2016, 48, 922–927. [Google Scholar] [CrossRef] [Green Version]
- Kastner, S.; Thiemann, I.-J.; Dekomien, G.; Petrasch-Parwez, E.; Schreiber, S.; Akkad, D.A.; Gerding, W.M.; Hoffjan, S.; Günes, S.; Günes, S.; et al. Exome sequencing reveals AGBL5 as novel candidate gene and additional variants for retinitis pigmentosa in five Turkish families. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8045–8053. [Google Scholar] [CrossRef] [Green Version]
- Astuti, G.D.N.; Arno, G.; Hull, S.; Pierrache, L.; Venselaar, H.; Carss, K.; Raymond, F.L.; Collin, R.W.J.; Faradz, S.M.H.; Born, L.I.V.D.; et al. Mutations in AGBL5, encoding a-tubulin deglutamylase, are associated with autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6180–6187. [Google Scholar] [CrossRef] [Green Version]
- Pathak, N.; Austin-Tse, C.A.; Liu, Y.; Vasilyev, A.; Drummond, A.I. Cytoplasmic carboxypeptidase 5 regulates tubulin glutamylation and zebrafish cilia formation and function. Mol. Biol. Cell 2014, 25, 1836–1844. [Google Scholar] [CrossRef]
- Vervoort, R.; Wright, A.F. Mutations of RPGR in X-linked retinitis pigmentosa (RP3). Hum. Mutat. 2002, 19, 486–500. [Google Scholar] [CrossRef]
- Koenekoop, R.K.; Loyer, M.; Hand, C.K.; Mahdi, H.A.; Dembinska, O.; Beneish, R.; Racine, J.; Rouleau, G.A. Novel RPGR mutations with distinct retinitis pigmentosa phenotypes in French-Canadian families. Am. J. Ophthalmol. 2003, 136, 678–687. [Google Scholar] [CrossRef]
- Hong, D.H.; Yue, G.; Adamian, M.; Li, T. Retinitis Pigmentosa GTPase Regulator (RPGR)-interacting Protein Is Stably Associated with the Photoreceptor Ciliary Axoneme and Anchors RPGR to the Connecting Cilium. J. Biol. Chem. 2001, 276, 12091–12099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, H. More than meets the eye: Current understanding of RPGR function. Adv. Exp. Med. Biol. 2018, 1074, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Mercado, C.L.; Pham, B.H.; Beres, S.; Marmor, M.F.; Lambert, S.R. Unilateral retinitis pigmentosa in children. J. AAPOS 2018, 22, 457–461. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Autoimmune Retinopathy/Cancer-Associated Retinopathy | Inflammatory Disease | Congenital Infection | Unilateral Retinitis Pigmentosa | Diagnosis |
|---|---|---|---|---|
| Positive history of systemic autoimmune diseases or neoplasms | History of eye redness or floaters or reduced vision | Maternal infection during pregnancy. Systemic manifestations of the disease | Positive family history can be found | History |
| Pigmentary retinopathy, can be normal examination | Anterior chamber or vitreous cells, anterior or posterior synechiae. Retinitis, vasculitis | Cataract, posterior synechiae, strabismus, retinal pigments | Unilateral bone spicules, attenuated vessels and pale optic disc | Clinical findings |
| Reduced photopic ± scotopic responses | Usually normal | Reduced photopic ± scotopic responses | Reduced scotopic ± photopic responses | Electroretinography |
| Positive antiretinal antibodies | Positive uveitis workup | Positive serologic testing for TORCH (toxoplasmosis, others (syphilis), rubella, cytomegalovirus, herpes) | Positive genetic testing | Lab/additional investigations |
| Patient 1 (Two Heterozygous Variants c.373 C>T (p.Arg125Trp) and c.730-22_730-19 dup in AGBL5 | Patient 2 (RPGR Missense Variant c.1286 C>T (p.Pro429Leu) | |||||||
|---|---|---|---|---|---|---|---|---|
| Year | VA OD | VA OS | CMT OD | CMT OS | VA OD | VA OS | CMT OD | CMT OS |
| 2015 | NA | NA | NA | NA | 20/20 | HM | 285 μm | 263 μm |
| 2016 | 20/20 | 20/40 | 287 μm | 285 μm | NA | NA | NA | NA |
| 2017 | 20/20 | 20/40 | 278 μm | 285 μm | NA | NA | NA | NA |
| 2018 | NA | NA | NA | NA | NA | NA | NA | NA |
| 2019 | 20/20 | 20/40 | 271 μm | 275 μm | 20/20 | HM | 274 μm | 255 μm |
| 2020 | NA | NA | NA | NA | 20/20 | HM | 262 μm | 245 μm |
| Authors | Age, Gender | Eye, Symptoms | Initial VA | Retinal Examination | ERG | VF | VA | Genetics |
|---|---|---|---|---|---|---|---|---|
| Makhupadhyay et al. 2011 [6] | 63, F | Right, asymptomatic | 20/30 | Bone spicules, attenuated vessels, pale disc | Flat scotopic and markedly reduced photopic | Mildly constricted | N/A | RP1 p.R677X heterozygous nonsense mutation |
| Marsiglia et al. 2013 [5] | 8, F | Right, nyctalopia | 20/20 | Bone spicules, attenuated vessels, pale disc | Markedly reduced scotopic and photopic | N/A | N/A | USH2A Trp4149Arg |
| Mercado et al. 2018 [25] | 15, F | Left, nyctalopia | 20/25 | Bone spicules, attenuated vessels, pale disc | Flat scotopic and photopic | N/A | 20/25 | USH2A gene c.6958-5 C>T (intronic splice variant) and c.6638T.A (Val2228Glu) (missense variant)) |
| Sim et al. 2018 [7] | 12, F | Right, asymptomatic | 6/5 | Bone spicules, attenuated vessels, pale disc | Flat scotopic and photopic | Severely constricted | 6/9 | CLRN1 heterozygous mutation c.118T>G (p.Cys40Gly) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milibari, D.; Magliyah, M.; Semidey, V.A.; Schatz, P.; ALBalawi, H.B. Unilateral Retinitis Pigmentosa Associated with Possible Ciliopathy and a Novel Mutation. Clin. Pract. 2022, 12, 491-500. https://doi.org/10.3390/clinpract12040053
Milibari D, Magliyah M, Semidey VA, Schatz P, ALBalawi HB. Unilateral Retinitis Pigmentosa Associated with Possible Ciliopathy and a Novel Mutation. Clinics and Practice. 2022; 12(4):491-500. https://doi.org/10.3390/clinpract12040053
Chicago/Turabian StyleMilibari, Doaa, Moustafa Magliyah, Valmore A. Semidey, Patrik Schatz, and Hani B. ALBalawi. 2022. "Unilateral Retinitis Pigmentosa Associated with Possible Ciliopathy and a Novel Mutation" Clinics and Practice 12, no. 4: 491-500. https://doi.org/10.3390/clinpract12040053
APA StyleMilibari, D., Magliyah, M., Semidey, V. A., Schatz, P., & ALBalawi, H. B. (2022). Unilateral Retinitis Pigmentosa Associated with Possible Ciliopathy and a Novel Mutation. Clinics and Practice, 12(4), 491-500. https://doi.org/10.3390/clinpract12040053