

Clinical Case of Mild Tatton–Brown–Rahman Syndrome Caused by a Nonsense Variant in DNMT3A Gene

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
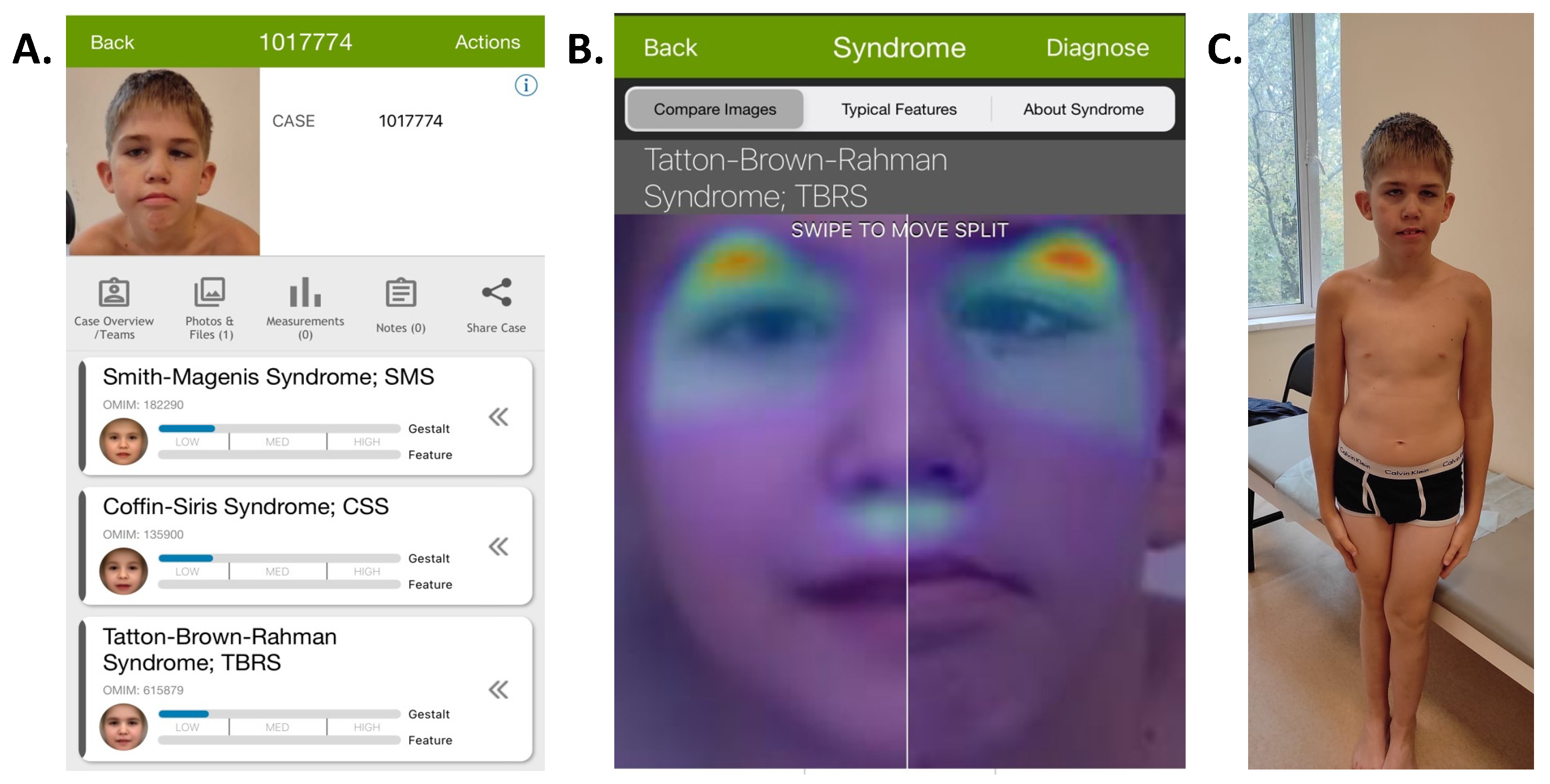
3.1. Case Description
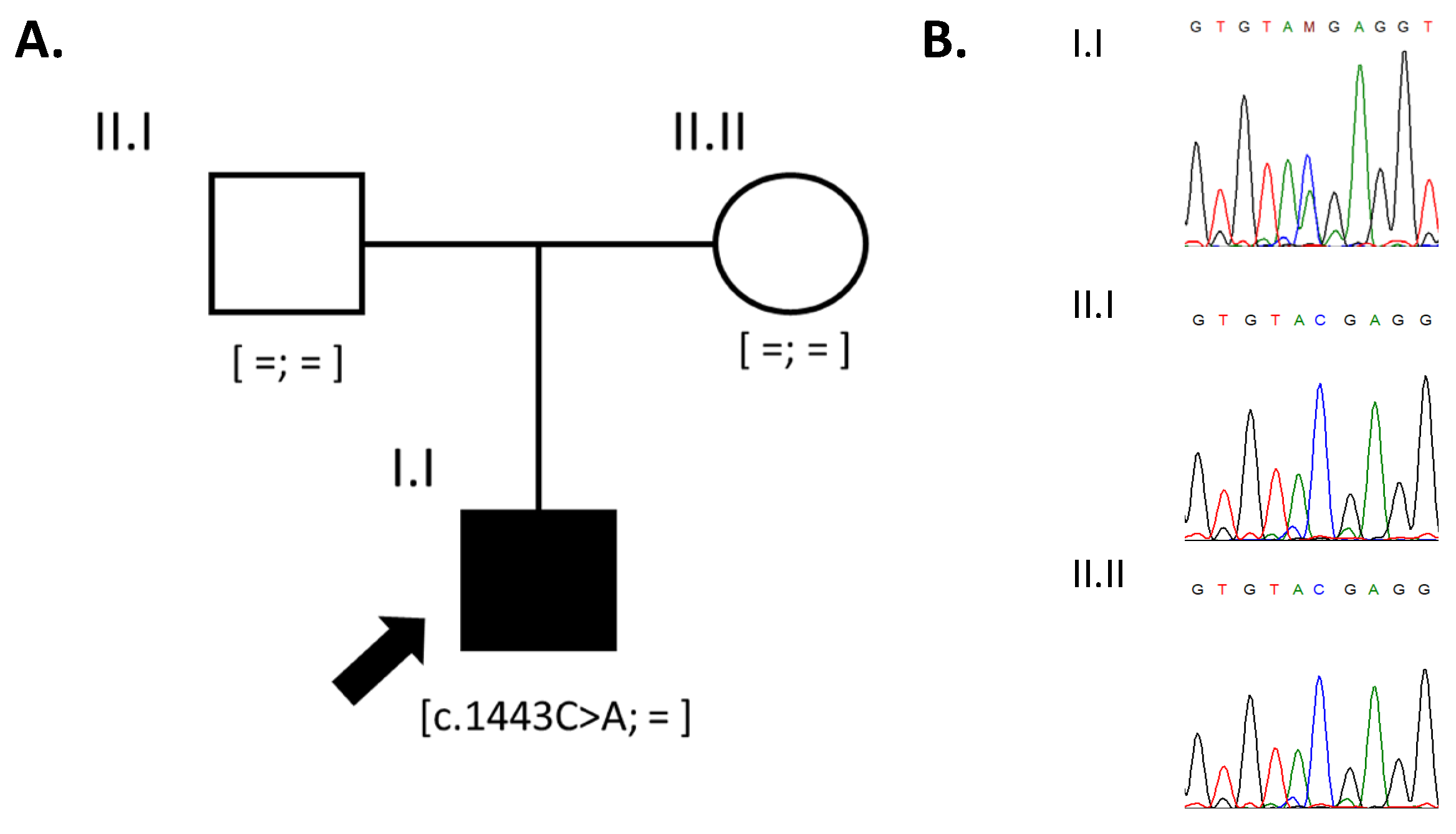
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tatton-Brown, K.; Seal, S.; Ruark, E.; Harmer, J.; Ramsay, E.; Del Vecchio Duarte, S.; Zachariou, A.; Hanks, S.; O’Brien, E.; Aksglaede, L.; et al. Mutations in the DNA Methyltransferase Gene DNMT3A Cause an Overgrowth Syndrome with Intellectual Disability. Nat. Genet. 2014, 46, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Structure and Function of Mammalian DNA Methyltransferases. Chembiochem 2011, 12, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Zachariou, A.; Loveday, C.; Renwick, A.; Mahamdallie, S.; Aksglaede, L.; Baralle, D.; Barge-Schaapveld, D.; Blyth, M.; Bouma, M.; et al. The Tatton-Brown-Rahman Syndrome: A Clinical Study of 55 Individuals with de Novo Constitutive DNMT3A Variants. Wellcome Open Res. 2018, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.; Cruz Marino, T.; Szekely, J.; Leblanc, J.; Cechner, K.; Sency, V.; Wensel, C.; Barabas, M.; Therriault, V.; Wang, H. Novel DNMT3A Germline Mutations Are Associated with Inherited Tatton-Brown-Rahman Syndrome. Clin. Genet. 2017, 91, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Hollink, I.H.I.M.; van den Ouweland, A.M.W.; Beverloo, H.B.; Arentsen-Peters, S.T.C.J.M.; Zwaan, C.M.; Wagner, A. Acute Myeloid Leukaemia in a Case with Tatton-Brown-Rahman Syndrome: The Peculiar DNMT3A R882 Mutation. J. Med. Genet. 2017, 54, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Tlemsani, C.; Luscan, A.; Leulliot, N.; Bieth, E.; Afenjar, A.; Baujat, G.; Doco-Fenzy, M.; Goldenberg, A.; Lacombe, D.; Lambert, L.; et al. SETD2 and DNMT3A Screen in the Sotos-like Syndrome French Cohort. J. Med. Genet. 2016, 53, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-J.; Xu, J.; Gu, Z.-H.; Pan, C.-M.; Lu, G.; Shen, Y.; Shi, J.-Y.; Zhu, Y.-M.; Tang, L.; Zhang, X.-W.; et al. Exome Sequencing Identifies Somatic Mutations of DNA Methyltransferase Gene DNMT3A in Acute Monocytic Leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A Mutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Levine, R.L. Mutations in Epigenetic Modifiers in the Pathogenesis and Therapy of Acute Myeloid Leukemia. Blood 2013, 121, 3563–3572. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Heeley, J.M.; Carlston, C.M.; Acuna-Hidalgo, R.; Nillesen, W.M.; Dent, K.M.; Douglas, G.V.; Levine, K.L.; Bayrak-Toydemir, P.; Marcelis, C.L.; et al. The Spectrum of DNMT3A Variants in Tatton-Brown-Rahman Syndrome Overlaps with That in Hematologic Malignancies. Am. J. Med. Genet. A 2017, 173, 3022–3028. [Google Scholar] [CrossRef] [PubMed]
- Heyn, P.; Logan, C.V.; Fluteau, A.; Challis, R.C.; Auchynnikava, T.; Martin, C.-A.; Marsh, J.A.; Taglini, F.; Kilanowski, F.; Parry, D.A.; et al. Gain-of-Function DNMT3A Mutations Cause Microcephalic Dwarfism and Hypermethylation of Polycomb-Regulated Regions. Nat. Genet. 2019, 51, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, O.; Dadali, E.; Bessonova, L.; Demina, N.; Rudenskaya, G.; Matyushchenko, G.; Markova, T.; Anisimova, I.; Semenova, N.; Shchagina, O.; et al. Complex Diagnostics of Non-Specific Intellectual Developmental Disorder. Int. J. Mol. Sci. 2022, 23, 7764. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Douglas, J.; Coleman, K.; Baujat, G.; Cole, T.R.P.; Das, S.; Horn, D.; Hughes, H.E.; Temple, I.K.; Faravelli, F.; et al. Genotype-Phenotype Associations in Sotos Syndrome: An Analysis of 266 Individuals with NSD1 Aberrations. Am. J. Hum. Genet. 2005, 77, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Murray, A.; Hanks, S.; Douglas, J.; Armstrong, R.; Banka, S.; Bird, L.M.; Clericuzio, C.L.; Cormier-Daire, V.; Cushing, T.; et al. Weaver syndrome and EZH2 mutations: Clarifying the clinical phenotype. Am. J. Med. Genet A 2013, 161A, 2972–2980. [Google Scholar] [CrossRef] [PubMed]
- Duffy, K.A.; Cielo, C.M.; Cohen, J.L.; Gonzalez-Gandolfi, C.X.; Griff, J.R.; Hathaway, E.R.; Kupa, J.; Taylor, J.A.; Wang, K.H.; Ganguly, A.; et al. Characterization of the Beckwith-Wiedemann Spectrum: Diagnosis and Management. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden Syndrome and the PTEN Hamartoma Tumor Syndrome: Systematic Review and Revised Diagnostic Criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Norio, R.; Raitta, C.; Lindahl, E. Further Delineation of the Cohen Syndrome; Report on Chorioretinal Dystrophy, Leukopenia and Consanguinity. Clin. Genet. 1984, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hersh, J.H.; Cole, T.R.; Bloom, A.S.; Bertolone, S.J.; Hughes, H.E. Risk of Malignancy in Sotos Syndrome. J. Pediatr. 1992, 120, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Cöktü, S.; Spix, C.; Kaiser, M.; Beygo, J.; Kleinle, S.; Bachmann, N.; Kohlschmidt, N.; Prawitt, D.; Beckmann, A.; Klaes, R.; et al. Cancer Incidence and Spectrum among Children with Genetically Confirmed Beckwith-Wiedemann Spectrum in Germany: A Retrospective Cohort Study. Br. J. Cancer 2020, 123, 619–623. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bostanova, F.; Levchenko, O.; Sharova, M.; Semenova, N. Clinical Case of Mild Tatton–Brown–Rahman Syndrome Caused by a Nonsense Variant in DNMT3A Gene. Clin. Pract. 2024, 14, 928-933. https://doi.org/10.3390/clinpract14030073
Bostanova F, Levchenko O, Sharova M, Semenova N. Clinical Case of Mild Tatton–Brown–Rahman Syndrome Caused by a Nonsense Variant in DNMT3A Gene. Clinics and Practice. 2024; 14(3):928-933. https://doi.org/10.3390/clinpract14030073
Chicago/Turabian StyleBostanova, Fatima, Olga Levchenko, Margarita Sharova, and Natalia Semenova. 2024. "Clinical Case of Mild Tatton–Brown–Rahman Syndrome Caused by a Nonsense Variant in DNMT3A Gene" Clinics and Practice 14, no. 3: 928-933. https://doi.org/10.3390/clinpract14030073