Bacterial Superantigen Toxins, CD28, and Drug Development

Abstract
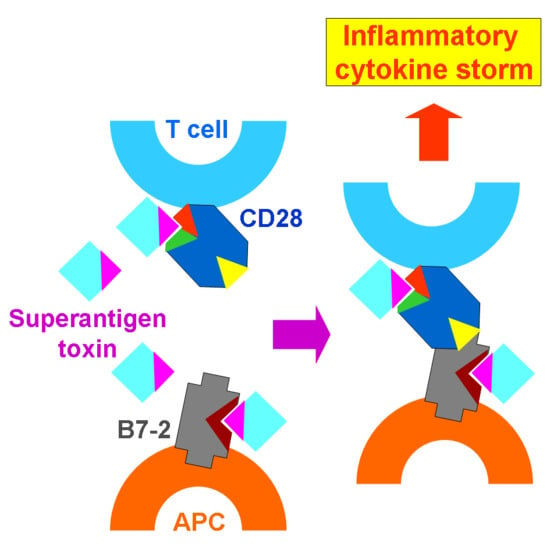
{kind=link}
{kind=link}
1. Introduction
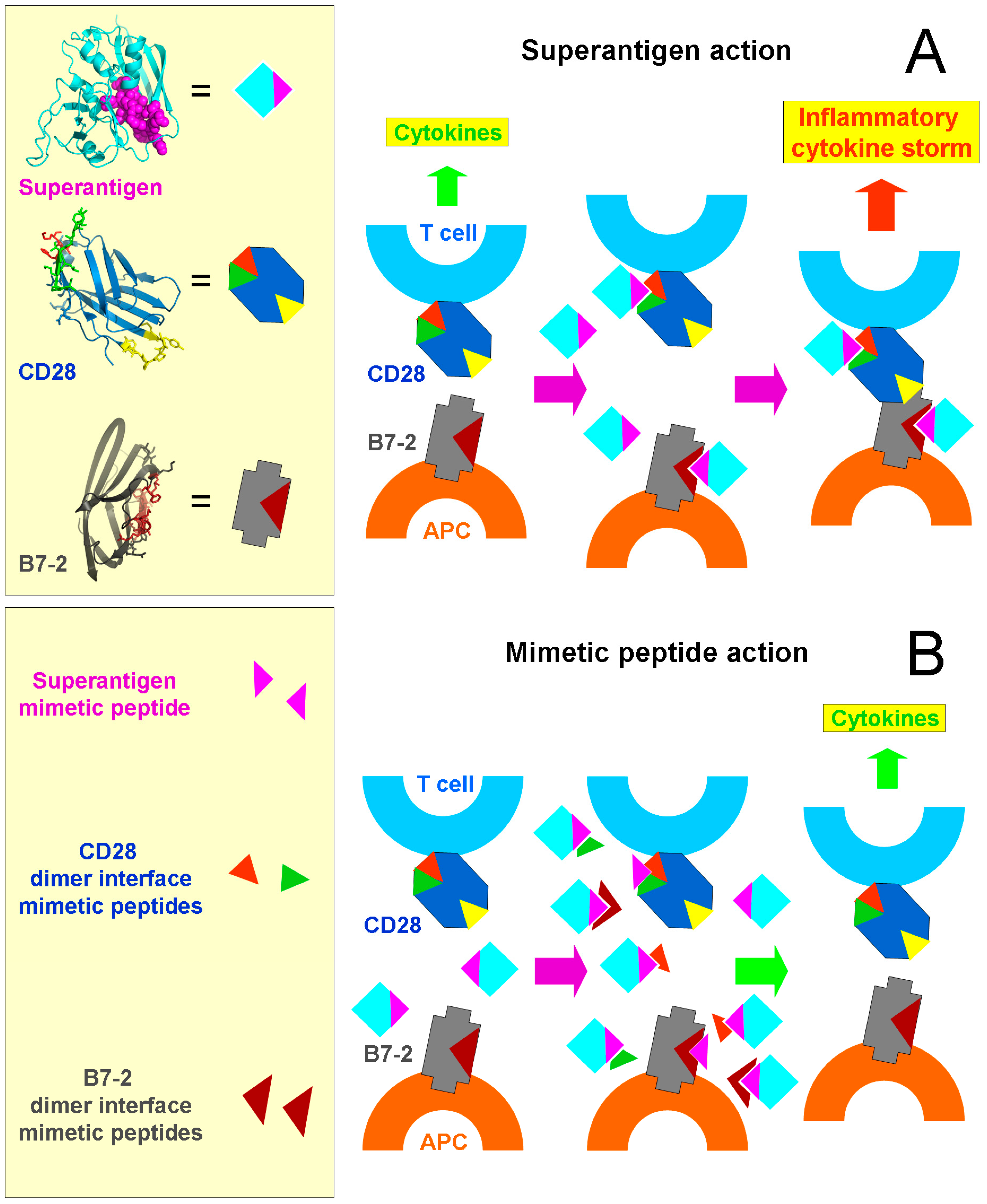
2. CD28 and B7-2 Are Key Superantigen Receptors and Dimer Interface Mimetic Peptides Protect from Death
3. The Superantigen Triggers B7-2/CD28 Costimulatory Receptor Engagement
4. Bacterial Infection
5. Conclusions
Funding
Conflicts of Interest
References
- Arad, G.; Levy, R.; Hillman, D.; Kaempfer, R. Superantigen antagonist protects against lethal shock and defines a new domain for T-cell activation. Nat. Med. 2000, 6, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Kaempfer, R.; Arad, G.; Levy, R.; Hillman, D. Defense against biologic warfare with superantigen toxins. Isr. Med. Assoc. J. 2002, 4, 520–523. [Google Scholar] [PubMed]
- United States Army Medical Research Institute of Infectious Diseases. Medical Management of Biological Casualties Handbook, 6th ed.; Woods, J.B., Ed.; United States Army Medical Research Institute of Infectious Diseases: Fort Detrick, MD, USA, 2005. Available online: http://www.dhhr.wv.gov/oeps/disease/Documents/USAMRIID_BlueBook.pdf (accessed on 6 November 2018).
- Marrack, P.; Blackman, M.; Kushnir, E.; Kappler, J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J. Exp. Med. 1990, 171, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: Critical role of tumor necrosis factor. J. Exp. Med. 1992, 175, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Leder, L.; Llera, A.; Lavoie, P.M.; Lebedeva, M.I.; Li, H.; Sékaly, R.P.; Bohach, G.A.; Gahr, P.J.; Schlievert, P.M.; Karjalainen, K.; et al. A mutational analysis of the binding of staphylococcal enterotoxins B and C3 to the T cell receptor beta chain and major histocompatibility complex class II. J. Exp. Med. 1998, 187, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of superantigen toxins into CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. 2011, 9, e1001149. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Rotfogel, Z.; Hillman, D.; Popugailo, A.; Arad, A.; Supper, E.; Osman, F.; Kaempfer, R. Superantigens hyperinduce inflammatory cytokines by enhancing the B7-2/CD28 costimulatory receptor interaction. Proc. Natl. Acad. Sci. USA 2016, 113, E6437–E6446. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L.; June, C.H. The CD28 family: A T-cell rheostat for therapeutic control of T-cell activation. Blood 2005, 105, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.; Kaempfer, R.; Chung, C.-S.; Chahin, A.B.; Palardy, J.E.; Parejo, N.A.; Chen, Y.; Whitford, M.; Arad, G.; Hillman, D.; et al. CD28 homodimer interface mimetic peptide acts as preventive and therapeutic agent in models of severe bacterial sepsis and gram-negative bacterial peritonitis. J. Infect. Dis. 2015, 211, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Bulger, E.M.; Maier, R.V.; Sperry, J.; Joshi, M.; Henry, S.; Moore, F.A.; Moldawer, L.L.; Demetriades, D.; Talving, P.; Schreiber, M.; et al. A novel drug for treatment of necrotizing soft-tissue infections: A randomized clinical trial. JAMA Surg. 2014, 149, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, T.; Lee, K.P.; Harris, E.S.; Petryniak, B.; Craighead, N.; Reynolds, P.J.; Lombard, D.B.; Freeman, G.J.; Nadler, L.M.; Gray, G.S.; et al. Characterization of CTLA-4 structure and expression on human T cells. J. Immunol. 1993, 151, 3489–3499. [Google Scholar] [PubMed]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Su, G.H.; Zuckerman, L.A.; Nabavi, N.; Jellis, C.L.; Gray, G.S.; Miller, J.; Bluestone, J.A. Expression and functional significance of an additional ligand for CTLA-4. Proc. Natl. Acad. Sci. USA 1993, 90, 11054–11058. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Edidin, M.; Almo, S.C.; Nathenson, S.G. B7-1 and B7-2: Similar costimulatory ligands with different biochemical, oligomeric and signaling properties. Immunol. Lett. 2006, 104, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Edidin, M.; Almo, S.C.; Nathenson, S.G. Different cell surface oligomeric states of B7-1 and B7-2: Implications for signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 15569–15574. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; Ikemizu, S.; Evans, E.J.; Fugger, L.; Bakker, T.R.; van der Merwe, P.A. The nature of molecular recognition by T cells. Nat. Immunol. 2003, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.J.; Esnouf, R.M.; Manso-Sancho, R.; Gilbert, R.J.; James, J.R.; Yu, C.; Fennelly, J.A.; Vowles, C.; Hanke, T.; Walse, B.; et al. Crystal structure of a soluble CD28-Fab complex. Nat. Immunol. 2005, 6, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Zhang, X.; Fedorov, A.A.; Nathenson, S.G.; Almo, S.C. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001, 410, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.; Tulapurkar, M.E.; Harris, K.M.; Arad, G.; Shirvan, A.; Shemesh, R.; DeTolla, L.J.; Benazzi, C.; Opal, S.M.; Kaempfer, R.; et al. A peptide antagonist of CD28 signaling attenuates toxic shock and necrotizing soft tissue infection induced by Streptococcus pyogenes. J. Infect. Dis. 2013, 207, 1869–1877. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaempfer, R. Bacterial Superantigen Toxins, CD28, and Drug Development. Toxins 2018, 10, 459. https://doi.org/10.3390/toxins10110459
Kaempfer R. Bacterial Superantigen Toxins, CD28, and Drug Development. Toxins. 2018; 10(11):459. https://doi.org/10.3390/toxins10110459
Chicago/Turabian StyleKaempfer, Raymond. 2018. "Bacterial Superantigen Toxins, CD28, and Drug Development" Toxins 10, no. 11: 459. https://doi.org/10.3390/toxins10110459
APA StyleKaempfer, R. (2018). Bacterial Superantigen Toxins, CD28, and Drug Development. Toxins, 10(11), 459. https://doi.org/10.3390/toxins10110459