Roles of Shiga Toxins in Immunopathology


{kind=link}
{kind=link}
Abstract
:1. Introduction
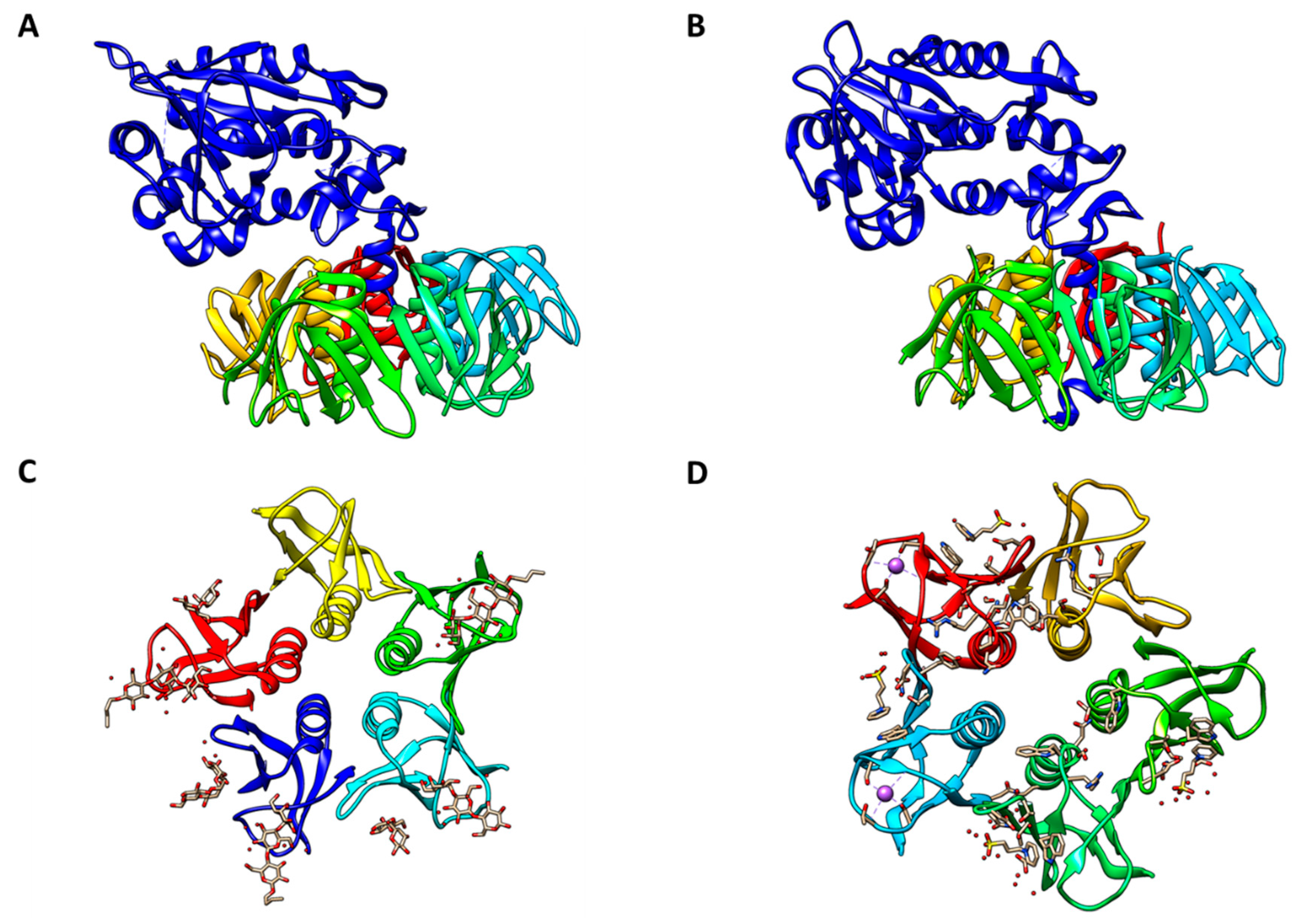
2. The Toxins
3. The Role of Stxs in Innate Immunity
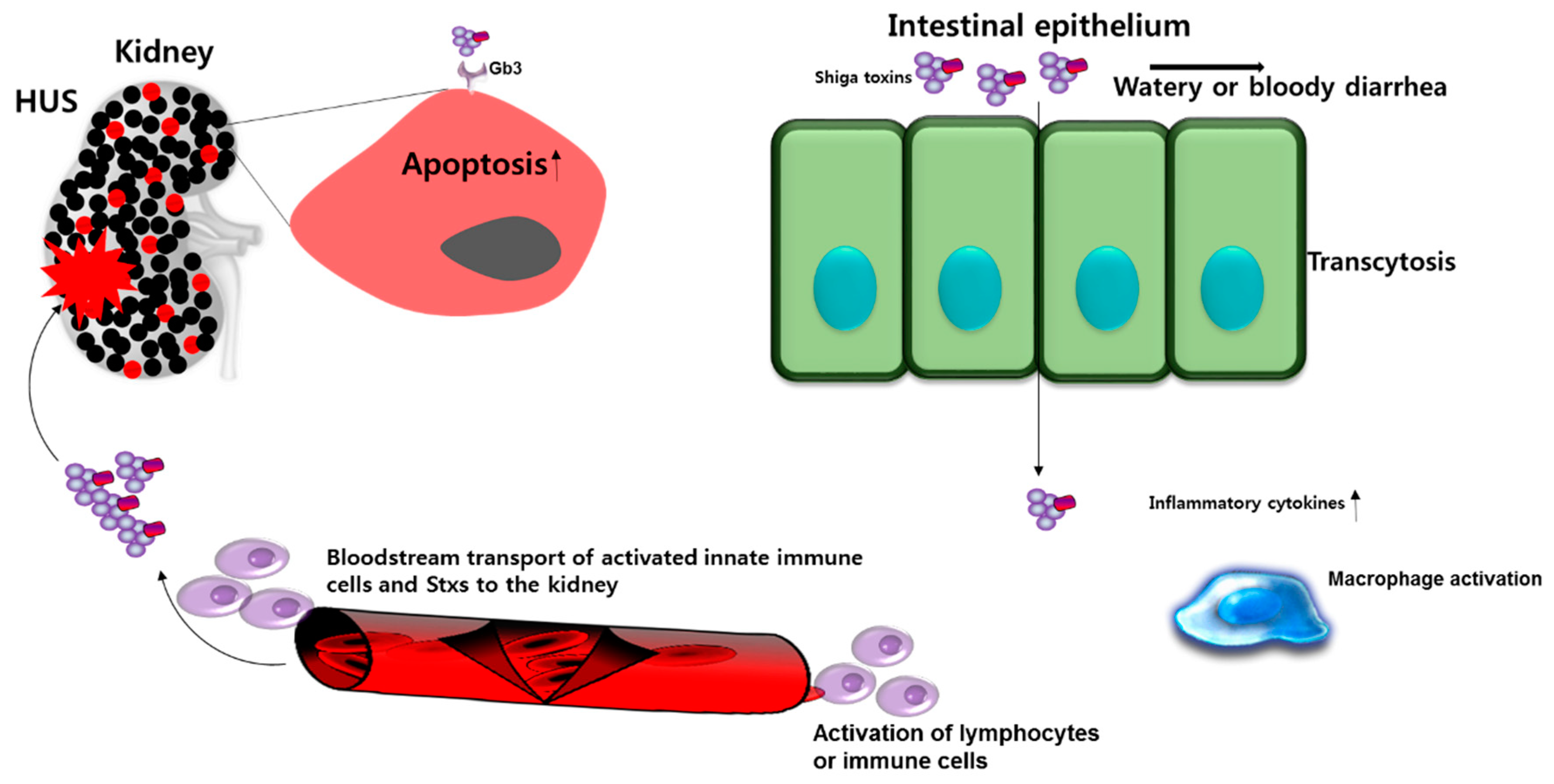
4. Role of Stxs in Gut Immunopathogenesis
5. The Role of Stxs in Renal Immunopathogenesis
6. Roles of Stxs in CNS Immunopathogenesis
7. The Immunopathological Role of Stxs in Circulating Blood During HUS Pathogenesis
8. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karmali, M.A. Emerging public health challenges of Shiga toxin-producing Escherichia coli related to changes in the pathogen, the population, and the environment. Clin. Infect. Dis. 2017, 64, 371–376. [Google Scholar] [PubMed]
- Kaper, J.B.; O’Brien, A.D. Overview and historical perspectives. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Parisot, M.; Parez, N.; Boukhari, R.; Breurec, S.; Jolivet, A. Shigella infection in children under 5 years old in Western French Guiana. Epidemiol. Infect. 2018, 146, 980–984. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Riddle, M.S.; Platts-Mills, J.A.; Pavlinac, P.; Zaidi, A.K.M. Shigellosis. Lancet 2018, 391, 801–812. [Google Scholar] [CrossRef]
- Gupta, S.K.; Strockbine, N.; Omondi, M.; Hise, K.; Fair, M.A.; Mintz, E. Emergence of Shiga toxin 1 genes within Shigella dysenteriae type 4 isolates from travelers returning from the Island of Hispañola. Am. J. Trop. Med. Hyg. 2007, 76, 1163–1165. [Google Scholar] [CrossRef] [PubMed]
- Beutin, L.; Strauch, E.; Fischer, I. Isolation of Shigella sonnei lysogenic for a bacteriophage encoding gene for production of Shiga toxin. Lancet 1999, 353, 1498. [Google Scholar] [CrossRef]
- Nógrády, N.; Király, M.; Borbás, K.; Tóth, Á.; Pászti, J.; Tóth, I. Antimicrobial resistance and genetic characteristics of integron-carrier Shigellae isolated in Hungary (1998-2008). J. Med. Microbiol. 2013, 62, 1545–1551. [Google Scholar] [CrossRef]
- Gray, M.D.; Lampel, K.A.; Strockbine, N.A.; Fernandez, R.E.; Melton-Celsa, A.R.; Maurelli, A.T. Clinical isolates of Shiga toxin 1a-producing Shigella flexneri with an epidemiological link to recent travel to Hispañiola. Emerg. Infect. Dis. 2014, 20, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, O.; Lienemann, T.; Halkilahti, J.; Mero, S.; Rimhanen-Finne, R.; Lehtinen, V.; Salmenlinna, S.; Siitonen, A. Characterization of Shigella sonnei isolate carrying Shiga toxin 2-producing gene. Emerg. Infect. Dis. 2015, 21, 891–892. [Google Scholar] [CrossRef]
- Lamba, K.; Nelson, J.A.; Kimura, A.C.; Poe, A.; Collins, J.; Kao, A.S.; Cruz, L.; Inami, G.; Vaishampayan, J.; Garz, A.; et al. Shiga toxin 1-producing Shigella sonnei infections, California, United States, 2014-2015. Emerg. Infect. Dis. 2016, 22, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Foodborne enterohemorrhagic Escherichia coli infections. In Preharvest and Postharvest Food Safety: Contemporary Issues and Future Directions; Beier, R.C., Pillai, S.D., Phillips, T.D., Ziprin, R.L., Eds.; Blackwell Publishing: Ames, IA, USA, 2004; Chapter 3; pp. 27–42. [Google Scholar]
- Heredia, N.; Garcia, S. Animals as sources of food-borne pathogens: A review. Anim. Nutr. 2018, 4, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Michino, H.; Araki, K.; Minami, S.; Takaya, S.; Sakai, N.; Miyazaki, M.; Ono, A.; Yanagawa, H. Massive outbreak of Escherichia coli O157:H7 infection in schoolchildren in Sakai City, Japan, associated with consumption of white radish sprouts. Am. J. Epidemiol. 1999, 150, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A. Host and pathogen determinants of verocytotoxin-producing Escherichia coli-associated hemolytic uremic syndrome. Kidney Int. 2009, 75 (Suppl. 112), S4–S7. [Google Scholar] [CrossRef]
- Scallan, E.; Mahon, B.E.; Hoekstra, R.M.; Griffin, P.M. Estimates of illnesses, hospitalizations and deaths caused by major bacterial enteric pathogens in young children in the United States. Pediatr. Infect. Dis. J. 2013, 32, 217–221. [Google Scholar] [CrossRef]
- Proulx, F.; Seidman, E.G.; Karpman, D. Pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Pediatr. Res. 2001, 50, 163–171. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Noris, M.; Remuzzi, G. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 2001, 60, 831–846. [Google Scholar] [CrossRef] [Green Version]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar] [CrossRef]
- Bruyand, M.; Mariani-Kurkdjian, P.; Gouali, M.; de Valk, H.; King, L.A.; Le Hello, S.; Bonacorsi, S.; Loirat, C. Hemolytic uremic syndrome due to Shiga toxin-producing Escherichia coli infection. Med. Mal. Infect. 2018, 48, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Ståhl, A.-L. Enterohemorrhagic Escherichia coli pathogenesis and the host response. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Lee, M.S.; Kwon, H.; Nguyen, L.T.; Lee, E.Y.; Lee, C.Y.; Choi, S.H.; Kim, M.H. Shiga toxins trigger the secretion of lysyl-tRNA synthetase to enhance proinflammatory responses. J. Microbiol. Biotechnol. 2016, 26, 432–439. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Park, S.K.; Yoon, S.J.; Park, Y.J.; Lee, M.S. Experimental in vivo models of bacterial Shiga toxin-associated hemolytic uremic syndrome. J. Microbiol. Biotechnol. 2018, 28, 1413–1425. [Google Scholar] [PubMed]
- Forsyth, K.D.; Fitzpatrick, M.M.; Simpson, A.C.; Barratt, T.M.; Levinsky, R.J. Neutrophil-mediated endothelial injury in haemolytic uraemic syndrome. Lancet 1989, 334, 411–414. [Google Scholar] [CrossRef]
- Morigi, M.; Micheletti, G.; Figliuzzi, M.; Imberti, B.; Karmali, M.A.; Remuzzi, A.; Remuzzi, G.; Zoja, C. Verotoxin-1 promotes leukocyte adhesion to cultured endothelial cells under physiologic flow conditions. Blood 1995, 86, 4553–4558. [Google Scholar] [PubMed]
- Lee, M.S.; Koo, S.; Jeong, D.G.; Tesh, V.L. Shiga toxins as multi-functional proteins: Induction of host cellular stress responses, role in pathogenesis and therapeutic applications. Toxins 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar] [PubMed]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 Å resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.D.; Armstrong, G.D.; Brunton, J.L.; Read, R.J. Structure of Shiga-like toxin I B-pentamer complexed with an analogue of its receptor Gb3. Biochemistry 1998, 37, 1777–1788. [Google Scholar] [CrossRef]
- Bast, D.J.; Banerjee, L.; Clark, C.; Read, R.J.; Brunton, J.L. The identification of three biologically relevant globotriaosyl ceramide receptor binding sites on the Verotoxin 1 B subunit. Mol. Microbiol. 1999, 32, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltyk, A.M.; MacKenzie, C.R.; Wolski, V.M.; Hirama, T.; Kitov, P.I.; Bundle, D.R.; Brunton, J.L. A mutational analysis of the globotriaosylceramide-binding sites of verotoxin VT1. J. Biol. Chem. 2002, 277, 5351–5359. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.P.; Neill, R.J.; O’Brien, A.D.; Holmes, R.K.; Newland, J.W. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiol. Lett. 1987, 44, 109–114. [Google Scholar] [CrossRef]
- Strockbine, N.A.; Marques, L.R.M.; Newland, J.W.; Williams Smith, H.; Holmes, R.K.; O’Brien, A.D. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immun. 1986, 53, 135–140. [Google Scholar]
- Scheutz, F.; Teel, L.D.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S.; et al. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R. Shiga toxin (Stx) classification, structure and function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Neely, M.N.; Friedman, D.I. Functional and genetic analysis of regulatory regions of coliphage H-19B: Location of Shiga-like toxin and lysis genes suggest a role for phage functions in toxin release. Mol. Microbiol. 1998, 28, 1255–1267. [Google Scholar] [CrossRef]
- Tyler, J.S.; Mills, M.J.; Friedman, D.I. The operator and early promoter region of the Shiga toxin type 2-encoding bacteriophage 933W and control of toxin expression. J. Bacteriol. 2004, 186, 7670–7679. [Google Scholar] [CrossRef] [PubMed]
- Łoś, J.M.; Łoś, M.; Węgrzyn, G. Bacteriophages carrying Shiga toxin genes: Genomic variations, detection and potential treatment of pathogenic bacteria. Future Microbiol. 2011, 6, 909–924. [Google Scholar] [CrossRef]
- Ostroff, S.M.; Tarr, P.I.; Neill, M.A.; Lewis, J.H.; Hargrett-Bean, N.; Kobayashi, J.M. Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 1989, 160, 994–998. [Google Scholar] [CrossRef] [PubMed]
- Boerlin, P.; McEwen, S.A.; Boerlin-Petzold, F.; Wilson, J.B.; Johnson, R.P.; Gyles, C.L. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. J. Clin. Microbiol. 1999, 37, 497–503. [Google Scholar] [PubMed]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [PubMed]
- Russo, L.M.; Melton-Celsa, A.R.; Smith, M.A.; Smith, M.J.; O’Brien, A.D. Oral intoxication of mice with Shiga toxin type 2a (Stx2a) and protection by anti-Stx2a monoclonal antibody 11E10. Infect. Immun. 2013, 82, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Jacewicz, M.; Clausen, H.; Nudelman, E.; Donohue-Rolfe, A.; Keusch, G.T. Pathogenesis of Shigella diarrhea. XI: Isolation of a Shigella toxin-binding glycolipid from rabbit jejunum and HeLa cells and its identification as globotriaosylceramide. J. Exp. Med. 1986, 163, 1391–1404. [Google Scholar] [CrossRef]
- Lindberg, A.A.; Brown, J.E.; Stromberg, N.; Westling-Ryd, M.; Schultz, J.E.; Karlsson, K.A. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J. Biol. Chem. 1987, 262, 1779–1785. [Google Scholar]
- Lingwood, C.A.; Law, H.; Richardson, S.; Petric, M.; Brunton, J.L.; DeGrandis, S.; Karmali, M. Glycolipid binding of purified and recombinant Escherichia coli produced verotoxin in vitro. J. Biol. Chem. 1987, 262, 8834–8839. [Google Scholar] [PubMed]
- Karve, S.S.; Weiss, A.A. Glycolipid binding preferences of Shiga toxin variants. PLoS ONE 2014, 9, e101173. [Google Scholar] [CrossRef]
- Legros, N.; Pohlentz, G.; Steil, D.; Kouzel, I.U.; Liashkovich, I.; Mellmann, A.; Karch, H.; Müthing, J. Membrane assembly of Shiga toxin glycosphingolipid receptors and toxin refractiveness of MDCK II epithelial cells. J. Lipid Res. 2018, 59, 1383–1401. [Google Scholar] [CrossRef] [PubMed]
- Müthing, J.; Meisen, I.; Zhang, W.; Bielaszewska, M.; Mormann, M.; Bauerfeind, R.; Schmidt, M.A.; Friedrich, A.W.; Karch, H. Promiscuous Shiga toxin 2e and its intimate relationship to Forssman. Glycobiology 2012, 22, 849–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steil, D.; Schepers, C.L.; Pohlentz, G.; Legros, N.; Runde, J.; Humpf, H.U.; Karch, H.; Müthing, J. Shiga toxin glycosphingolipid receptors of Vero-B4 kidney epithelial cells and their membrane microdomain lipid environment. J. Lipid Res. 2015, 56, 2322–2336. [Google Scholar] [CrossRef] [PubMed]
- Kiarash, A.; Boyd, B.; Lingwood, C.A. Glycosphingolipid receptor function is modified by fatty acid content. Verotoxin 1 and verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J. Biol. Chem. 1994, 269, 11138–11346. [Google Scholar]
- Arab, S.; Lingwood, C.A. Influence of phospholipid chain length on verotoxin/globotriaosyl ceramide binding in model membranes: Comparison of a supported bilayer film and liposomes. Glycoconj. J. 1996, 13, 159–166. [Google Scholar] [CrossRef]
- Binnington, B.; Lingwood, D.; Nutikka, A.; Lingwood, C.A. Effect of globotriaosyl ceramide fatty acid α-hydroxylation on the binding by Verotoxin 1 and Verotoxin 2. Neurochem. Res. 2002, 27, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Watkins, E.B.; Gao, H.; Dennison, A.J.; Chopin, N.; Struth, B.; Arnold, T.; Florent, J.C.; Johannes, L. Carbohydrate conformation and lipid condensation in monolayers containing glycosphingolipid Gb3: Influence of acyl chain structure. Biophys. J. 2014, 107, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Proulx, F.; Lingwood, C.A. Detergent-resistant globotriaosyl ceramide may define verotoxin/glomeruli-restricted hemolytic uremic syndrome pathology. Kidney Int. 2009, 75, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfoud, R.; Manis, A.; Binnington, B.; Ackerley, C.; Lingwood, C.A. A major fraction of glycosphingolipids in model and cellular cholesterol-containing membranes is undetectable by their binding proteins. J. Biol. Chem. 2010, 285, 36049–36059. [Google Scholar] [CrossRef]
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.; Fraisier, V.; Florent, J.C.; Perrais, D.; et al. Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 2007, 450, 670–675. [Google Scholar] [CrossRef]
- Sandvig, K.; Olsnes, S.; Brown, J.E.; Petersen, O.W.; van Deurs, B. Endocytosis from coated pits of Shiga toxin: A glycolipid-binding protein from Shigella dysenteriae 1. J. Cell Biol. 1989, 108, 1331–1343. [Google Scholar] [CrossRef]
- Lauvrak, S.U.; Torgersen, M.L.; Sandvig, K. Efficient endosome-to-Golgi transport of Shiga toxin is dependent on dynamin and clathrin. J. Cell Sci. 2004, 117, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Saint-Pol, A.; Yélamos, B.; Amessou, M.; Mills, I.G.; Dugast, M.; Tenza, D.; Schu, P.; Antony, C.; McMahon, H.T.; Lamaze, C.; et al. Clathrin adaptor epsinR is required for retrograde sorting on early endosomal membranes. Dev. Cell 2004, 6, 525–538. [Google Scholar] [CrossRef]
- Sandvig, K.; Kavaliauskiene, S.; Skotland, T. Clathrin-independent endocytosis: An increasing degree of complexity. Histochem. Cell Biol. 2018, 150, 107–118. [Google Scholar] [CrossRef]
- Johannes, L. Shiga toxin—A model for glycolipid-dependent and lectin-driven endocytosis. Toxins 2017, 9, 340. [Google Scholar] [CrossRef]
- Pezeshkian, W.; Hansen, A.G.; Johannes, L.; Khandelia, H.; Shillcock, J.; Sunil Kumar, P.B.; Ipsen, J.H. Membrane invagination induced by Shiga toxin B-subunit: From molecular structure to tube formation. Soft Matter 2016, 12, 5164–5171. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Garred, Ø.; Prydz, K.; Kozlov, J.V.; Hansen, S.H.; van Deurs, B. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 1992, 358, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Garred, Ø.; van Deurs, B.; Sandvig, K. Furin-induced cleavage and activation of Shiga toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Reisbig, R.; Eiklid, K. Subunit structure of Shigella cytotoxin. J. Biol. Chem. 1981, 256, 8732–8738. [Google Scholar] [PubMed]
- Spooner, R.A.; Lord, J.M. How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 2012, 357, 19–40. [Google Scholar] [PubMed]
- Correll, C.C.; Munishkin, A.; Chan, Y.L.; Ren, Z.; Wool, I.G.; Steitz, T.A. Crystal structure of the ribosomal RNA domain essential for binding elongation factors. Proc. Natl. Acad. Sci. USA 1998, 95, 13436–13441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumer, N.E.; Li, X.-P. Interaction of ricin and Shiga toxins with ribosomes. Curr. Topics Microbiol. Immunol. 2012, 357, 1–18. [Google Scholar]
- Shi, W.-W.; Nga-Sze, A.; Wong, K.-B.; Shaw, P.-C. Structures and ribosomal interaction of ribosome-inactivating proteins. Molecules 2016, 21, 1588. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef]
- La Pointe, P.; Wei, X.; Gariepy, J. A role for the protease-sensitive loop region of Shiga-like toxin 1 in the retrotranslocation of its A1 domain from the endoplasmic reticulum lumen. J. Biol. Chem. 2005, 280, 23310–23318. [Google Scholar] [CrossRef] [PubMed]
- Adnan, H.; Zhang, Z.; Park, H.-J.; Tailor, C.; Che, C.; Kamani, M.; Spitalny, G.; Binnington, B.; Lingwood, C. Endoplasmic reticulum-targeted subunit toxins provide a new approach to rescue misfolded mutant proteins and revert cell models of genetic diseases. PLoS ONE 2016, 11, e0166948. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Immunol. Rev. 2009, 8, 958–969. [Google Scholar] [CrossRef]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesh, V.L.; Ramegowda, B.; Samuel, J.E. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect. Immun. 1994, 62, 5085–5094. [Google Scholar] [PubMed]
- Brandelli, J.R.; Griener, T.P.; Laing, A.; Mulvey, G.; Armstrong, G.D. The effects of Shiga toxin 1, 2 and their subunits on cytokine and chemokine expression by human macrophage-like THP-1 cells. Toxins 2015, 7, 4054–4066. [Google Scholar] [CrossRef]
- Leyva-Illades, D.; Cherla, R.P.; Lee, M.S.; Tesh, V.L. Regulation of cytokine and chemokine expression by the ribotoxic stress response elicited by Shiga toxin type 1 in human macrophage-like THP-1 cells. Infect. Immun. 2012, 80, 2109–2120. [Google Scholar] [CrossRef]
- Legros, N.; Pohlentz, G.; Steil, D.; Müthing, J. Shiga toxin-glycosphingolipid interaction: Status quo of research with focus on primary human brain and kidney endothelial cells. Int. J. Med. Microbiol. 2018, 308, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Ebel, F.; Podzadel, T.; Rohde, M.; Kresse, A.U.; Kramer, S.; Deibel, C.; Guzman, C.A.; Chakraborty, T. Initial binding of Shiga toxin-producing Escherichia coli to host cells and subsequent induction of actin rearrangements depend on filamentous EspA-containing surface appendages. Mol. Microbiol. 1998, 30, 147–161. [Google Scholar] [CrossRef]
- Wang, H.; Rogers, T.J.; Paton, J.C.; Paton, A.W. Differential effects of Escherichia coli subtilase cytotoxin and Shiga toxin 2 on chemokine and proinflammatory cytokine expression in human macrophage, colonic epithelial, and brain microvascular endothelial cell lines. Infect. Immun. 2014, 82, 3567–3579. [Google Scholar] [CrossRef]
- Keepers, T.R.; Gross, L.K.; Obrig, T.G. Monocyte chemoattractant protein 1, macrophage inflammatory protein 1α, and RANTES recruit macrophages to the kidney in a mouse model of hemolytic-uremic syndrone. Infect. Immun. 2007, 75, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kwon, H.; Lee, E.Y.; Kim, D.J.; Park, J.H.; Tesh, V.L.; Oh, T.K.; Kim, M.H. Shiga toxins activate the NLRP3 inflammasome pathway to promote both production of the proinflammatory cytokine interleukin-1beta and apoptotic cell death. Infect. Immun. 2016, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Wadolkowski, E.A.; Sung, L.M.; Burris, J.A.; Samuel, J.E.; O’Brien, A.D. Acute renal tubular necrosis and death of mice orally infected with Escherichia coli strains that produce Shiga-like toxin type II. Infect. Immun. 1990, 58, 3959–3965. [Google Scholar] [PubMed]
- Shibolet, O.; Shina, A.; Rosen, S.; Cleary, T.G.; Brezis, M.; Ashkenazi, S. Shiga toxin induces medullary tubular injury in isolated perfused rat kidneys. FEMS Immunol. Med. Microbiol. 1997, 18, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, F.B., Jr.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.K.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the baboon responses to Shiga-like toxin: Descriptive study of a new primate model of toxic responses to Stx-1. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar] [CrossRef]
- Hughes, A.K.; Stricklett, P.K.; Kohan, D.E. Cytotoxic effect of Shiga toxin-1 on human proximal tubule cells. Kidney Int. 1998, 54, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Foster, G.H.; Bitzan, M. Silencing of Bak ameliorates apoptosis of human proximal tubular epithelial cells by Escherichia coli-derived Shiga toxin 2. Infection 2005, 33, 362–367. [Google Scholar] [CrossRef]
- Lentz, E.K.; Leyva-Illades, D.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Differential response of the human renal proximal tubular epithelial cell line HK-2 to Shiga toxin types 1 and 2. Infect. Immun. 2011, 79, 3527–3540. [Google Scholar] [CrossRef]
- Porubsky, S.; Federico, G.; Müthing, J.; Jennemann, R.; Gretz, N.; Büttner, S.; Obermüller, N.; Jung, O.; Hauser, I.A.; Gröne, E.; et al. Direct acute tubular damage contributes to Shigatoxin-mediated kidney failure. J. Pathol. 2014, 234, 120–133. [Google Scholar] [CrossRef] [Green Version]
- Sansonetti, P.J. Rupture, invasion and inflammatory destruction of the intestinal barrier by Shigella, making sense of prokaryote-eukaryote cross-talks. FEMS Microbiol. Rev. 2001, 25, 3–14. [Google Scholar] [CrossRef]
- Mellouk, N.; Enninga, J. Cytosolic access of intracellular bacterial pathogens: The Shigella paradigm. Front. Cell Infect. Microbiol. 2016, 6, 35. [Google Scholar] [CrossRef]
- Fontaine, A.; Arondel, J.; Sansonetti, P.J. Role of Shiga toxin in the pathogenesis of bacillary dysentery studied by using a Tox− mutant of Shigella dysenteriae 1. Infect. Immun. 1988, 56, 3099–3109. [Google Scholar]
- Phillips, A.D.; Navabpour, S.; Hicks, S.; Dougan, G.; Wallis, T.; Frankel, G. Enterohaemorrhagic Escherichia coli O157:H7 target Peyer’s patches in humans and cause attaching/effacing lesions in both human and bovine intestine. Gut 2000, 47, 377–381. [Google Scholar] [CrossRef]
- Chong, Y.; Fitzhenry, R.; Heuschkel, R.; Torrente, F.; Frankel, G.; Phillips, A.D. Human intestinal tissue tropism in Escherichia coli O157:H7—Initial colonization of terminal ileum and Peyer’s patches and minimal colonic adhesion ex vivo. Microbiology 2007, 153, 794–802. [Google Scholar] [CrossRef]
- Xicohtencatl-Cortes, J.; Monteiro-Neto, V.; Ledesma, M.A.; Jordan, D.M.; Francetic, O.; Kaper, J.B.; Puente, J.L.; Girón, J.A. Intestinal adherence associated with type IV pili of enterohemorrhagic Escherichia coli O157:H7. J. Clin. Investig. 2007, 117, 3519–3529. [Google Scholar] [CrossRef]
- Knutton, S.; Baldwin, T.; Williams, P.H.; McNeish, A.S. Actin accumulation at sites of bacterial adhesion to tissue culture cells: Basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 1987, 57, 1290–1298. [Google Scholar]
- McDaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 1995, 92, 1664–1668. [Google Scholar] [CrossRef]
- Campellone, K.G.; Leong, J.M. Tails of two Tirs: Actin pedestal formation by enteropathogenic E. coli and enterohemorrhagic E. coli O157:H7. Curr. Opin. Microbiol. 2003, 6, 82–90. [Google Scholar] [CrossRef]
- Furniss, R.C.D.; Clements, A. Regulation of the locus of enterocyte effacement in attaching and effacing pathogens. J. Bacteriol. 2018, 200, e00336-17. [Google Scholar] [CrossRef]
- Gaytán, M.O.; Martinez-Santos, V.I.; Soto, E.; González-Pedrajo, B. Type three secretion systems in attaching and effacing pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 129. [Google Scholar] [CrossRef]
- Hughes, D.T.; Clarke, M.B.; Yamamoto, K.; Rasko, D.A.; Sperandio, V. The QseC adrenergic signaling cascade in enterohemorrhagic E. coli (EHEC). PLoS Pathog. 2009, 5, e1000553. [Google Scholar] [CrossRef]
- Moreira, C.G.; Russell, R.; Mishra, A.A.; Narayanan, S.; Ritchie, J.M.; Waldor, M.K.; Curtis, M.M.; Winter, S.E.; Weinshenker, D.; Sperandio, V. Bacterial adrenergic sensors regulate virulence of enteric pathogens in the gut. mBio 2016, 7, e00826-16. [Google Scholar] [CrossRef]
- Mallick, E.M.; Garber, J.J.; Vanguri, V.K.; Balasubramanian, S.; Blood, T.; Clark, S.; Vingadassalom, D.; Louissaint, C.; McCormick, B.; Snapper, S.B.; et al. The ability of an attaching and effacing pathogen to trigger localized actin assembly contributes to virulence by promoting mucosal attachment. Cell. Microbiol. 2014, 16, 1405–1424. [Google Scholar] [CrossRef] [Green Version]
- Ugalde-Silva, P.; Gonzalez-Lugo, O.; Navarro-Garcia, F. Tight junction disruption induced by type 3 secretion system effectors injected by enteropathogenic and enterohemorrhagic Escherichia coli. Front. Cell. Infect. Microbiol. 2016, 6, 87. [Google Scholar] [CrossRef]
- Thorpe, C.M.; Hurley, B.P.; Lincicome, L.L.; Jacewicz, M.S.; Keusch, G.T.; Acheson, D.W. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999, 67, 5985–5993. [Google Scholar]
- Thorpe, C.M.; Smith, W.E.; Hurley, B.P.; Acheson, D.W. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 2001, 69, 6140–6147. [Google Scholar] [CrossRef]
- Guo, M.; Yang, W.; Wu, F.; Hao, G.; Li, R.; Wang, X.; Wei, L.; Chai, T. Colonization, mortality, and host cytokines response to enterohemorrhagic Escherichia coli in rabbits. Oncotarget 2017, 8, 93426–93437. [Google Scholar] [CrossRef]
- Yamasaki, C.; Natori, Y.; Zeng, X.T.; Ohmura, M.; Yamasaki, S.; Takeda, Y.; Natori, Y. Induction of cytokines in a human colon epithelial cell line by Shiga toxin 1 (Stx1) and Stx2 but not by non-toxic mutant Stx1 which lacks N-glycosidase activity. FEBS Lett. 1999, 442, 231–234. [Google Scholar] [CrossRef]
- Kelly, J.; Oryshak, A.; Wenetsek, M.; Grabiec, J.; Handy, S. The colonic pathology of Escherichia coli O157:H7 infection. Am. J. Surg. Pathol. 1990, 14, 87–92. [Google Scholar] [CrossRef]
- Békássy, Z.D.; Calderon-Toledo, C.; Leoj, G.; Kristoffersson, A.C.; Perez, M.-T.; Karpman, D. Intestinal damage in enterohemorrhagic Escherichia coli infection. Pediatr. Nephrol. 2011, 26, 2059–2071. [Google Scholar] [CrossRef]
- Heyderman, R.S.; Soriani, M.; Hirst, T.R. Is immune cell activation the missing link in the pathogenesis of post-diarrhoeal HUS? Trends Microbiol. 2001, 9, 262–266. [Google Scholar] [CrossRef]
- Lo, D.D. Vigilance or subversion? Constitutive and inducible M cells in mucosal tissues. Trends Immunol. 2018, 39, 185–195. [Google Scholar] [CrossRef]
- Etienne-Mesmin, L.; Chassaing, B.; Sauvanet, P.; Denizot, J.; Blanquet-Diot, S.; Darfeuille-Michaud, A.; Pradel, N.; Livrelli, V. Interactions with M cells and macrophages as key steps in the pathogenesis of enterohemorrhagic Escherchia coli infections. PLoS ONE 2011, 6, e23594. [Google Scholar] [CrossRef]
- Acheson, D.W.; Moore, R.; DeBreucker, S.; Lincicome, L.; Jacewicz, M.; Skutelsky, E.; Keusch, G.T. Translocation of Shiga toxin across polarized intestinal cells in tissue culture. Infect. Immun. 1996, 64, 3294–3300. [Google Scholar]
- Lukyanenko, V.; Malyukova, I.; Hubbard, A.; Delannoy, M.; Boedeker, E.; Zhu, C.; Cebotaru, L.; Kovbasnjuk, O. Enterohemorrhagic Escherichia coli infection stimulates Shiga toxin 1 macropinocytosis and transcytosis across intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2011, 301, C1140–C1149. [Google Scholar] [CrossRef]
- In, J.; Lukyanenko, V.; Foulke-Abel, J.; Hubbard, A.L.; Delannoy, M.; Hansen, A.M.; Kaper, J.B.; Boisen, N.; Nataro, J.P.; Zhu, C.; et al. Serine protease EspP from enterohemorrhagic Escherichia coli is sufficient to induce Shiga toxin macropinocytosis in intestinal epithelium. PLoS ONE 2013, 8, e69196. [Google Scholar] [CrossRef]
- Hurley, B.P.; Thorpe, C.M.; Acheson, D.W. Shiga toxin translocation across intestinal epithelial cells is enhanced by neutrophil transmigration. Infect. Immun. 2001, 69, 6148–6155. [Google Scholar] [CrossRef]
- Viswanathan, V.K.; Koutsouris, A.; Lukic, S.; Pilkinton, M.; Simonovic, I.; Simonovic, M.; Hecht, G. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect. Immun. 2004, 72, 3218–3227. [Google Scholar] [CrossRef]
- Schüller, S.; Frankel, G.; Phillips, A.D. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell. Microbiol. 2004, 6, 289–301. [Google Scholar] [CrossRef]
- Kovbasnjuk, O.; Mourtazina, R.; Baibakov, B.; Wang, T.; Elowsky, C.; Choti, M.A.; Kane, A.; Donowitz, M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 19087–19092. [Google Scholar] [CrossRef] [Green Version]
- Falguières, T.; Maak, M.; von Weyhern, C.; Sarr, M.; Sastre, X.; Poupon, M.F.; Robine, S.; Johannes, L.; Janssen, K.P. Human colorectal tumors and metastases express Gb3 and can be targeted by an intestinal pathogen-based delivery tool. Mol. Cancer Ther. 2008, 7, 2498–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumbrun, S.D.; Hanson, L.; Sinclair, J.F.; Freedy, J.; Melton-Celsa, A.R.; Rodriguez-Canales, J.; Hanson, J.C.; O’Brien, A.D. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mRNA and the alternate Shiga toxin receptor globotetraosylceramide. Infect. Immun. 2010, 78, 4488–4499. [Google Scholar] [CrossRef] [PubMed]
- Schüller, S.; Heuschkel, R.; Torrente, F.; Kaper, J.B.; Phillips, A.D. Shiga toxin binding in normal and inflamed human intestinal mucosa. Microb. Infect. 2007, 9, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.R.; Lazarus, J.E.; Sit, B.; Schmieder, S.; Lencer, W.I.; Blondel, C.J.; Doench, J.G.; Davis, B.M.; Waldor, M.K. CRISPR screen reveals that EHEC’s T3SS and Shiga toxin rely on shared host factors for infection. mBio 2018, 9, e01003-18. [Google Scholar] [CrossRef] [PubMed]
- Battle, S.E.; Brady, M.J.; Vanaja, S.K.; Leong, J.M.; Hecht, G.A. Actin pedestal formation by enterohemorrhagic Escherichia coli enhances bacterial host cell attachment and concomitant Type III translocation. Infect. Immun. 2014, 82, 3713–3722. [Google Scholar] [CrossRef]
- Campellone, K.G. Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia coli: Tir, EspFu and actin pedestal assembly. FEBS J. 2010, 277, 2390–2402. [Google Scholar] [CrossRef]
- Tian, S.; Muneeruddin, K.; Choi, M.Y.; Tao, L.; Bhuiyan, R.H.; Ohmi, Y.; Furukawa, K.; Furukawa, K.; Boland, S.; Shaffer, S.A.; et al. Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation. PLoS Biol. 2018, 16, e2006951. [Google Scholar] [CrossRef]
- Potelle, S.; Morelle, W.; Dulary, E.; Duvet, S.; Vicogne, D.; Spriet, C.; Krzewinski-Recchi, M.-A.; Morsomme, P.; Jaeken, J.; Matthijs, G.; et al. Glycosylation abnormalities in Gdt1p/TMEM165 deficient cells result from a defect in Golgi manganese homeostasis. Hum. Mol. Genet. 2016, 25, 1489–1500. [Google Scholar] [CrossRef] [Green Version]
- Louise, C.B.; Obrig, T.G. Shiga toxin-associated hemolytic-uremic syndrome: Combined cytotoxic effects of Shiga toxin, interleukin-1 beta, and tumor necrosis factor alpha on human vascular endothelial cell in vitro. Infect. Immun. 1991, 59, 4173–4179. [Google Scholar]
- van de Kar, N.C.A.J.; Monnens, L.A.H.; Karmali, M.A.; van Hinsbergh, V.W.M. Tumor necrosis factor and interleukin 1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: Implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar]
- Ramegowda, B.; Samuel, J.E.; Tesh, V.L. Interaction of Shiga toxins with human brain microvascular endothelial cells: Cytokines as sensitizing agents. J. Infect. Dis. 1999, 180, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Jacewicz, M.S.; Acheson, D.W.; Binion, D.G.; West, G.A.; Lincicome, L.L.; Fiocchi, C.; Keusch, G.T. Responses of human intestinal microvascular endothelial cells to Shiga toxins 1 and 2 and pathogenesis of hemorrhagic colitis. Infect. Immun. 1999, 67, 1439–1444. [Google Scholar] [PubMed]
- Ramegowda, B.; Tesh, V.L. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity to Shiga-like toxins in human monocytes and monocytic cell lines. Infect. Immun. 1996, 64, 1173–1180. [Google Scholar] [PubMed]
- Harrison, L.M.; van Haaften, W.C.E.; Tesh, V.L. Regulation of proinflammatory cytokine expression by Shiga toxin 1 and/or lipopolysaccharide in the human monocytic cell line THP-1. Infect. Immun. 2004, 72, 2618–2627. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; Cherla, R.P.; van den Hoogen, C.; van Haaften, W.C.E.; Lee, S.-Y.; Tesh, V.L. Comparative evaluation of apoptosis induced by Shiga toxin 1 and/or lipopolysaccharides in human monocytic and macrophage-like cells. Microb. Pathog. 2005, 38, 63–76. [Google Scholar] [CrossRef]
- Villysson, A.; Tontanahal, A.; Karpman, D. Microvesicle involvement in Shiga toxin-associated infection. Toxins 2017, 9, 376. [Google Scholar] [CrossRef]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Tamassia, N.; Borsetti, F.; Fabbri, E.; Tazzari, P.L.; Ricci, F.; Pagliaro, P.; Spisni, E.; et al. Identification of TLR4 as the receptor that recognizes Shiga toxins in human neutrophils. J. Immunol. 2013, 191, 4748–4758. [Google Scholar] [CrossRef]
- Brigotti, M. The interactions of human neutrophils with Shiga toxins and related plant toxins: Danger or safety? Toxins 2012, 4, 157–190. [Google Scholar] [CrossRef]
- Ståhl, A.-L.; Arvidsson, I.; Johansson, K.E.; Chromek, M.; Rebetz, J.; Loos, S.; Kristoffersson, A.-C.; Békássy, Z.D.; Mörgelin, M.; Karpman, D. A novel mechanism of bacterial toxin transfer within host blood cell-derived microvesicles. PLoS Pathog. 2015, 11, e1004619. [Google Scholar] [CrossRef]
- Schüller, S. Shiga toxin interaction with human intestinal epithelium. Toxins 2011, 3, 626–639. [Google Scholar] [CrossRef]
- Matussek, A.; Lauber, J.; Bergau, A.; Hansen, W.; Rohde, M.; Dittmar, K.E.; Gunzer, M.; Mengel, M.; Gatzlaff, P.; Hartmann, M. Molecular and functional analysis of Shiga toxin-induced response patterns in human vascular endothelial cells. Blood 2003, 102, 1323–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.K.; Stricklett, P.K.; Kohan, D.E. Shiga toxin-1 regulation of cytokine production by human proximal tubule cells. Kidney Int. 1998, 54, 1093–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic uraemic syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [CrossRef]
- Exeni, R.A.; Fernandez-Brando, R.J.; Santiago, A.P.; Fiorentino, G.A.; Exeni, A.M.; Ramos, M.V.; Palermo, M.S. Pathogenic role of inflammatory response during Shiga toxin-associated hemolytic uremic syndrome (HUS). Pediatr. Nephrol. 2018, 33, 2057–2071. [Google Scholar] [CrossRef]
- Lentz, E.K.; Cherla, R.P.; Jaspers, V.; Weeks, B.R.; Tesh, V.L. Role of tumor necrosis factor alpha in disease using a mouse model of Shiga toxin-mediated renal damage. Infect. Immun. 2010, 78, 3689–3699. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Oh, S.Y.; Cherla, R.P.; Lee, M.-S.; Tesh, V.L.; Papin, J.; Henderson, J.; Kurosawa, S. Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic Escherichia coli Shiga toxins in non-human primate models of hemolytic uremic syndrome. Am. J. Pathol. 2013, 182, 1227–1238. [Google Scholar] [CrossRef]
- de Lind van Wijngaarden, R.A.; Hauer, H.A.; Wolterbeek, R.; Jayne, D.R.; Gaskin, G.; Rasmussen, N.; Noel, L.H.; Ferrario, F.; Waldherr, R.; Hagen, E.C.; et al. Clinical and histologic determinants of renal outcome in ANCA-associated vasculitis: A prospective analysis of 100 patients with severe renal involvement. J. Am. Soc. Nephrol. 2006, 17, 2264–2274. [Google Scholar] [CrossRef]
- Bitzan, M.; Moebius, E.; Ludwig, K.; Müller-Wiefel, D.E.; Heesemann, J.; Karch, H. High incidence of serum antibodies to Escherichia coli O157 lipopolysaccharide in children with hemolytic-uremic syndrome. J. Pediatr. 1991, 119, 380–385. [Google Scholar] [CrossRef]
- Greatorex, J.S.; Thorne, G.M. Humoral immune responses to Shiga-like toxins and Escherichia coli O157 lipopolysaccharide in hemolytic-uremic syndrome patients and healthy subjects. J. Clin. Microbiol. 1994, 32, 1172–1178. [Google Scholar]
- Morrison, D.C.; Silverstein, R.; Luchi, M.; Shnyra, A. Structure-function relationships of bacterial endotoxins—Contribution to microbial sepsis. Infect. Dis. Clin. N. Am. 1999, 13, 313–340. [Google Scholar] [CrossRef]
- Sakiri, R.; Ramegowda, B.; Tesh, V.L. Shiga toxin type 1 activates tumor necrosis factor-alpha gene transcription and nuclear translocation of the transcriptional activators nuclear factor-κB and activator protein-1. Blood 1998, 92, 558–566. [Google Scholar]
- Cameron, P.; Bingham, D.; Paul, A.; Pavelka, M.; Cameron, S.; Rotondo, D.; Plevin, R. Essential role for verotoxin in sustained stress-activated protein kinase and nuclear factor kappa B signaling, stimulated by Escherichia coli O157:H7 in Vero cells. Infect. Immun. 2002, 70, 5370–5380. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Angioletti, S.; Donadelli, R.; Zanchi, C.; Tomasoni, S.; Binda, E.; Imberti, B.; te Loo, M.; Monnens, L.; Remuzzi, G.; et al. Shiga toxin 2 triggers endothelial leukocyte adhesion and transmigration via NF-kappaB dependent up-regulation of IL-8 and MCP-1. Kidney Int. 2002, 62, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Kim, M.H.; Tesh, V.L. Shiga toxins expressed by human pathogenic bacteria induce immune responses in host cells. J. Microbiol. 2013, 51, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.V.; Ruggieri, M.; Panek, A.C.; Mejias, M.P.; Fernandez-Brando, R.J.; Abrey-Recalde, M.J.; Exeni, A.; Barilari, C.; Exeni, R.; Palermo, M.S. Association of haemolytic uraemic syndrome with dysregulation of chemokine receptor expression in circulating monocytes. Clin. Sci. 2015, 129, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Grif, K.; Khan, A.B.; Naim, A.; Dierich, M.P.; Würzner, R. The Shiga toxin genotype rather than the amount of Shiga toxin or cytotoxicity of Shiga toxin in vitro correlates with the appearance of the hemolytic uremic syndrome. Diagn. Microbiol. Infect. Dis. 2007, 59, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.S.; Mooney, J.C.; Brandt, J.R.; Staples, A.O.; Jelacic, S.; Boster, D.R.; Watkins, S.L.; Tarr, P.I. Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: A multivariate analysis. Clin. Infect. Dis. 2012, 55, 33–41. [Google Scholar] [CrossRef]
- Tarr, G.A.; Oltean, H.N.; Phipps, A.I.; Rabinowitz, P.; Tarr, P.I. Strength of the association between antibiotic use and hemolytic uremic syndrome following Escherichia coli O157:H7 infection varies with case definition. Int. J. Med. Microbiol. 2018, 308, 921–926. [Google Scholar] [CrossRef]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of Shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar] [CrossRef]
- Rohde, H.; Qin, J.; Cui, Y.; Li, D.; Loman, N.J.; Hentschke, M.; Chen, W.; Pu, F.; Peng, Y.; Li, J.; et al. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N. Engl. J. Med. 2011, 365, 718–724. [Google Scholar] [CrossRef]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.A.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmanm, A.; Zhang, W.; Köck, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterization of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: A microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef]
- Magnus, T.; Röther, J.; Simova, O.; Meier-Cillien, M.; Repenthin, J.; Möller, F.; Gbadamosi, J.; Panzer, U.; Wengenroth, M.; Hagel, C.; et al. The neurological syndrome in adults during the 2011 northern German E. coli serotype O104:H4 outbreak. Brain 2012, 135, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Gould, L.H.; Demma, L.; Jones, T.F.; Hurd, S.; Vugia, D.J.; Smith, K.; Shiferaw, B.; Segler, S.; Palmer, A.; Zansky, S.; et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin. Infect. Dis. 2009, 49, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Heiman, K.E.; Mody, R.K.; Johnson, S.D.; Griffin, P.M.; Gould, L.H. Escherichia coli O157:H7 outbreaks in the United States, 2003-2012. Emerg. Infect. Dis. 2015, 21, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, P.; Lan, R.; Ye, C.; Wang, H.; Ren, J.; Jing, H.; Wang, Y.; Zhou, Z.; Bai, X.; et al. A novel Escherichia coli O157:H7 clone causing a major hemolytic uremic syndrome outbreak in China. PLoS ONE 2012, 7, e36144. [Google Scholar] [CrossRef]
- Harrison, L.M.; Gaines, D.W.; Babu, U.S.; Balan, K.V.; Reimschuessel, R.; Do, A.B.; Pereira, M.R.; Bigley, E.C., 3rd; Ferguson, M.; Mehta, A.; et al. Diet-induced obesity precipitates kidney dysfunction and alters inflammatory mediators in mice treated with Shiga toxin 2. Microb. Pathog. 2018, 123, 250–258. [Google Scholar] [CrossRef]
- Geelen, J.M.; van der Velden, T.J.; van den Heuvel, L.P.; Monnens, L.A. Interactions of Shiga-like toxin with human peripheral blood monocytes. Pediatr. Nephrol. 2007, 22, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Cooling, L.L.; Walker, K.E.; Gille, T.; Koerner, T.A. Shiga toxin binds human platelets via globotriaosylceramide (Pk antigen) and a novel platelet glycosphingolipid. Infect. Immun. 1998, 66, 4355–4366. [Google Scholar]
- Ghosh, S.A.; Polanowska-Grabowska, R.K.; Fujii, J.; Obrig, T.; Gear, A.R. Shiga toxin binds to activated platelets. J. Thromb. Haemost. 2004, 2, 499–506. [Google Scholar] [CrossRef]
- Bitzan, M.; Richardson, S.; Huang, C.; Boyd, B.; Petric, M.; Karmali, M.A. Evidence that verotoxins (Shiga-like toxins) from Escherichia coli bind to P blood group antigens of human erythrocytes in vitro. Infect. Immun. 1994, 62, 3337–3347. [Google Scholar] [PubMed]
- Betz, J.; Dorn, I.; Kouzel, I.U.; Bauwens, A.; Meisen, I.; Kemper, B.; Bielaszewska, M.; Mormann, M.; Weymann, L.; Sibrowski, W.; et al. Shiga toxin of enterohaemorrhagic Escherichia coli directly injures developing human erythrocytes. Cell. Microbiol. 2016, 18, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Monnens, L.; Molenaar, J.; Lambert, P.H.; Proesmans, W.; van Munster, P. The complement system in hemolytic-uremic syndrome in childhood. Clin. Nephrol. 1980, 13, 168–171. [Google Scholar] [PubMed]
- Keir, L.S.; Saleem, M.A. Current evidence for the role of complement in the pathogenesis of Shiga toxin haemolytic uraemic syndrome. Pediatr. Nephrol. 2014, 29, 1895–1902. [Google Scholar] [CrossRef]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative pathway activation of complement by Shiga toxin promotes exuberant C3a formation that triggers microvascular thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef]
- Locatelli, M.; Buelli, S.; Pezzotta, A.; Corna, D.; Perico, L.; Tomasoni, S.; Rottoli, D.; Rizzo, P.; Conti, D.; Thurman, J.M.; et al. Shiga toxin promotes podocyte injury in experimental hemolytic uremic syndrome via activation of the alternative pathway of complement. J. Am. Soc. Nephrol. 2014, 25, 1786–1798. [Google Scholar] [CrossRef] [PubMed]
- Tryggvason, K. Unraveling the mechanisms of glomerular ultrafiltration: Nephrin, a key component of the slit diaphragm. J. Am. Soc. Nephrol. 1999, 10, 2440–2445. [Google Scholar]
- Kretzler, M.; Teixeira, V.P.; Unschuld, P.G.; Cohen, C.D.; Wanke, R.; Edenhofer, I.; Mundel, P.; Schlondorff, D.; Holthofer, H. Integrin-linked kinase as a candidate downstream effector in proteinuria. FASEB J. 2001, 15, 1843–1845. [Google Scholar] [CrossRef]
- Kang, Y.S.; Li, Y.; Dai, C.; Kiss, L.P.; Wu, C.; Liu, Y. Inhibition of integrin-linked kinase blocks podocyte epithelial-mesenchymal transition and ameliorates proteinuria. Kidney Int. 2010, 78, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Khan, A.B.; Naim, A.; Grif, K.; Brockmeyer, J.; Karch, H.; Joannidis, M.; Clark, S.J.; Day, A.J.; Fidanzi, S.; et al. Shiga toxin activates complement and binds factor H: Evidence for an active role of complement in hemolytic uremic syndrome. J. Immunol. 2009, 182, 6394–6400. [Google Scholar] [CrossRef] [PubMed]
- Poolpol, K.; Orth-Holler, D.; Speth, C.; Zipfel, P.F.; Skerka, C.; de Cordoba, S.R.; Brockmeyer, J.; Bielaszewska, M.; Wurzner, R. Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol. Immunol. 2014, 58, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Trachtman, H.; Austin, C.; Lewinski, M.; Stahl, R.A. Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat. Rev. Nephrol. 2012, 8, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Sheth, K.J.; Swick, H.M.; Haworth, N. Neurological involvement in hemolytic-uremic syndrome. Ann. Neurol. 1986, 19, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Loos, S.; Wehrmann, C.; Horstmann, D.; Donnerstag, F.; Lemke, J.; Hillebrand, G.; Lobel, U.; Pape, L.; Haffner, D.; et al. Neurological involvement in children with E. coli O104:H4-induced hemolytic uremic syndrome. Pediatr. Nephrol. 2014, 29, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, S.; Kwon, T.; Elmaleh, M.; Charbit, M.; Launay, E.A.; Harambat, J.; Brun, M.; Ranchin, B.; Bandin, F.; Cloarec, S.; et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin. J. Am. Soc. Nephrol. 2010, 5, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Sahin, S.; Ozdogan, E.B.; Kaya, G.; Ozgun, N.; Cansu, A.; Kalyoncu, M.; Dilber, E. Neurological involvement in pediatric hemolytic uremic syndrome: A symptom-oriented analysis. Neuropediatrics 2017, 48, 363–370. [Google Scholar] [PubMed]
- Loudon, S.E.; Dorresteijn, E.M.; Catsman-Berrevoets, C.E.; Verdijk, R.M.; Simonsz, H.J.; Jansen, A.J. Blinded by Shiga toxin-producing O104 Escherichia coli and hemolytic uremic syndrome. J. Pediatr. 2014, 165, 410–410.e1. [Google Scholar] [CrossRef]
- Park, J.Y.; Jeong, Y.J.; Park, S.K.; Yoon, S.J.; Choi, S.; Jeong, D.G.; Chung, S.W.; Lee, B.J.; Kim, J.H.; Tesh, V.L.; et al. Shiga toxins induce apoptosis and ER stress in human retinal pigment epithelial cells. Toxins 2017, 9, 319. [Google Scholar] [CrossRef]
- Siegler, R.L.; Obrig, T.G.; Pysher, T.J.; Tesh, V.L.; Denkers, N.D.; Taylor, F.B., Jr. Response to Shiga toxin 1 and 2 in a baboon model of hemolytic uremic syndrome. Pediatr. Nephrol. 2003, 18, 92–96. [Google Scholar]
- Siegler, R.L.; Pysher, T.J.; Tesh, V.L.; Taylor, F.B., Jr. Response to single and divided doses of Shiga toxin-1 in a primate model of hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2001, 12, 1458–1467. [Google Scholar]
- Pozhilenkova, E.A.; Lopatina, O.L.; Komleva, Y.K.; Salmin, V.V.; Salmina, A.B. Blood-brain barrier-supported neurogenesis in healthy and diseased brain. Rev. Neurosci. 2017, 28, 397–415. [Google Scholar] [CrossRef]
- Legros, N.; Dusny, S.; Humpf, H.-U.; Pohlentz, G.; Karch, H.; Müthing, J. Shiga toxin glycosphingolipid receptors and their lipid membrane ensemble in primary human blood-brain barrier endothelial cells. Glycobiology 2017, 27, 99–109. [Google Scholar] [CrossRef]
- Lingwood, C.A.; Binnington, B.; Manis, A.; Branch, D.R. Globotriaosyl ceramide receptor function—Where membrane structure and pathology intersect. FEBS Lett. 2010, 584, 1879–1886. [Google Scholar] [CrossRef]
- Eisenhauer, P.B.; Jacewicz, M.S.; Conn, K.J.; Koul, O.; Wells, J.M.; Fine, R.E.; Newburg, D.S. Escherichia coli Shiga toxin 1 and TNF-alpha induce cytokine release by human cerebral microvascular endothelial cells. Microb. Pathog. 2004, 36, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landoni, V.I.; de Campos-Nebel, M.; Schierloh, P.; Calatayud, C.; Fernandez, G.C.; Ramos, M.V.; Rearte, B.; Palermo, M.S.; Isturiz, M.A. Shiga toxin 1-induced inflammatory response in lipopolysaccharide-sensitized astrocytes is mediated by endogenous tumor necrosis factor alpha. Infect. Immun. 2010, 78, 1193–1201. [Google Scholar] [CrossRef]
- Landoni, V.I.; Schieroh, P.; de Campos Nebel, M.; Fernández, G.C.; Calatayud, C.; Lapponi, M.J.; Isturiz, M.A. Shiga toxin 1 induces on lipopolysaccharide-treated astrocytes the release of tumor necrosis factor-alpha that alter brain-like endothelium integrity. PLoS Pathog. 2012, 8, e1002632. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Funata, N.; Ikuta, F.; Sato, S. Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with Shiga toxin-2. J. Neuroinflamm. 2008, 5, 11. [Google Scholar] [CrossRef]
- Pinto, A.; Cangelosi, A.; Geoghegan, P.A.; Goldstein, J. Dexamethasone prevents motor deficits and neurovascular damage produced by Shiga toxin 2 and lipopolysaccharide in the mouse striatum. Neuroscience 2017, 344, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Meuth, S.G.; Göbel, K.; Kanyshkova, T.; Ehling, P.; Ritter, M.A.; Schwindt, W.; Bielaszewska, M.; Lebiedz, P.; Coulon, P.; Herrmann, A.M.; et al. Thalamic involvement in patients with neurologic impairment due to Shiga toxin 2. Ann. Neurol. 2012, 73, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Obrig, T.G.; Louise, C.B.; Lingwood, C.A.; Boyd, B.; Barley-Maloney, L.; Daniel, T.O. Endothelial heterogeneity in Shiga toxin receptors and responses. J. Biol. Chem. 1993, 268, 15484–15488. [Google Scholar] [PubMed]
- Te Loo, D.M.; van Hinsbergh, V.W.; van den Heuvel, L.P.; Monnens, L.A. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2001, 12, 800–806. [Google Scholar] [PubMed]
- Brigotti, M.; Caprioli, A.; Tozzi, A.E.; Tazzari, P.L.; Ricci, F.; Conte, R.; Carnicelli, D.; Procaccino, M.A.; Minelli, F.; Ferretti, A.V.; et al. Shiga toxins present in the gut and in the polymorphonuclear leukocytes circulating in the blood of children with hemolytic-uremic syndrome. J. Clin. Microbiol. 2006, 44, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Te Loo, D.M.; Monnens, L.A.; van der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; van den Heuvel, L.P.; van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar] [PubMed]
- Torgersen, M.L.; Engedal, N.; Pedersen, A.-M.; Husebye, H.; Espevik, T.; Sandvig, K. Toll-like receptor 4 facilitates binding of Shiga toxin to colon carcinoma and primary umbilical vein endothelial cells. FEMS Immunol. Med. Microbiol. 2011, 61, 63–75. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Lauvrak, S.U.; Sandvig, K. The A-subunit of surface-bound Shiga toxin stimulates clathrin-dependent uptake of the toxin. FEBS J. 2005, 272, 4103–4113. [Google Scholar] [CrossRef] [Green Version]
- Griener, T.P.; Mulvey, G.L.; Marcato, P.; Armstrong, G.D. Differential binding of Shiga toxin 2 to human and murine neutrophils. J. Med. Microbiol. 2007, 56, 1423–1430. [Google Scholar] [CrossRef] [Green Version]
- Arfilli, V.; Carnicelli, D.; Rocchi, L.; Ricci, F.; Pagliaro, P.; Tazzari, P.L.; Brigotti, M. Shiga toxin 1 and ricin A chain bind to human polymorphonuclear leucocytes through a common receptor. Biochem. J. 2010, 432, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Porcellini, E.; Galassi, E.; Valerii, M.C.; Spisni, E. Human monocytes stimulated by Shiga toxin 1a via globotriaosylceramide release proinflammatory molecules associated with hemolytic uremic syndrome. Int. J. Med. Microbiol. 2018, 308, 940–946. [Google Scholar] [CrossRef]
- Niu, S.; Paluszynski, J.; Bian, Z.; Shi, L.; Kidder, K.; Liu, Y. LPS-primed CD11b(+) leukocytes serve as an effective carrier of Shiga toxin 2 to cause hemolytic uremic syndrome in mice. Sci. Rep. 2018, 8, 3994. [Google Scholar] [CrossRef]
- van de Kar, N.C.; Kooistra, T.; Vermeer, M.; Lesslauer, W.; Monnens, L.A.; van Hinsbergh, V.W. Tumor necrosis factor alpha induces endothelial galactosyl transferase activity and verocytotoxin receptors. Role of specific tumor necrosis factor receptors and protein kinase C. Blood 1995, 85, 734–743. [Google Scholar] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.-S.; Tesh, V.L. Roles of Shiga Toxins in Immunopathology. Toxins 2019, 11, 212. https://doi.org/10.3390/toxins11040212
Lee M-S, Tesh VL. Roles of Shiga Toxins in Immunopathology. Toxins. 2019; 11(4):212. https://doi.org/10.3390/toxins11040212
Chicago/Turabian StyleLee, Moo-Seung, and Vernon L. Tesh. 2019. "Roles of Shiga Toxins in Immunopathology" Toxins 11, no. 4: 212. https://doi.org/10.3390/toxins11040212
APA StyleLee, M.-S., & Tesh, V. L. (2019). Roles of Shiga Toxins in Immunopathology. Toxins, 11(4), 212. https://doi.org/10.3390/toxins11040212