Separation and Purification of Aflatoxins by Centrifugal Partition Chromatography

 , and
, and 
Abstract
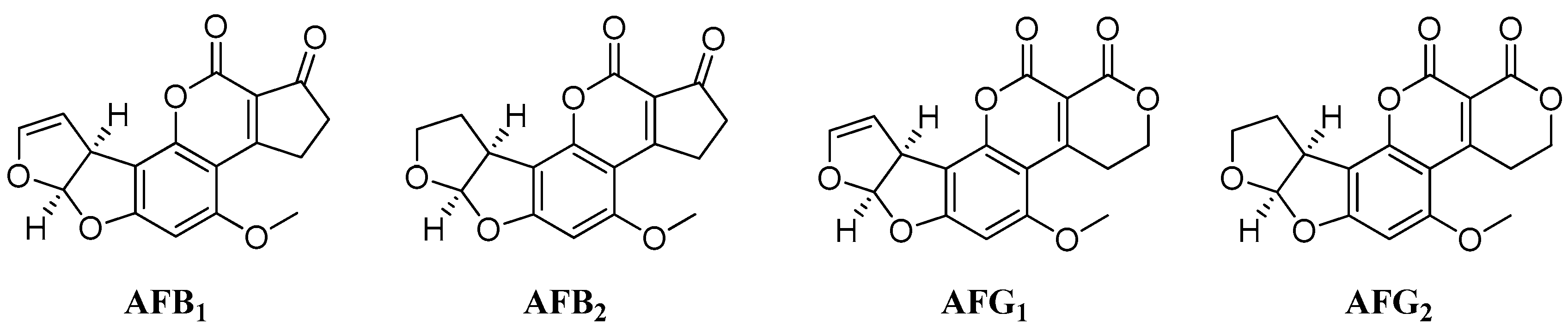
:1. Introduction
2. Results and Discussion
2.1. Cultivation and Extraction
2.2. Selection of Solvent Systems
2.2.1. Chloroform and Acetone as Best Solvents
2.2.2. Acetic Acid as Best Solvent
2.3. Optimisation of the CPC Method
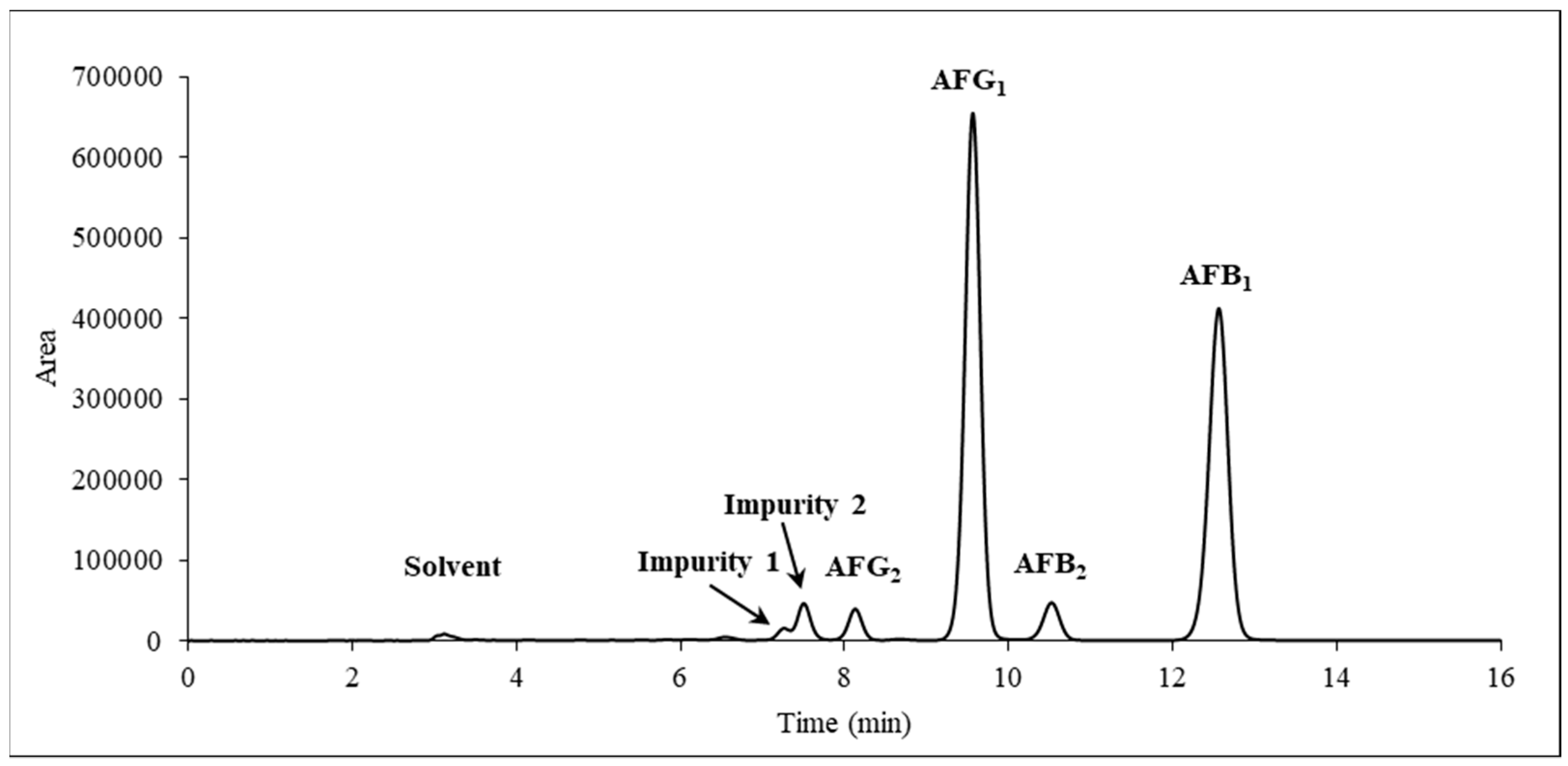
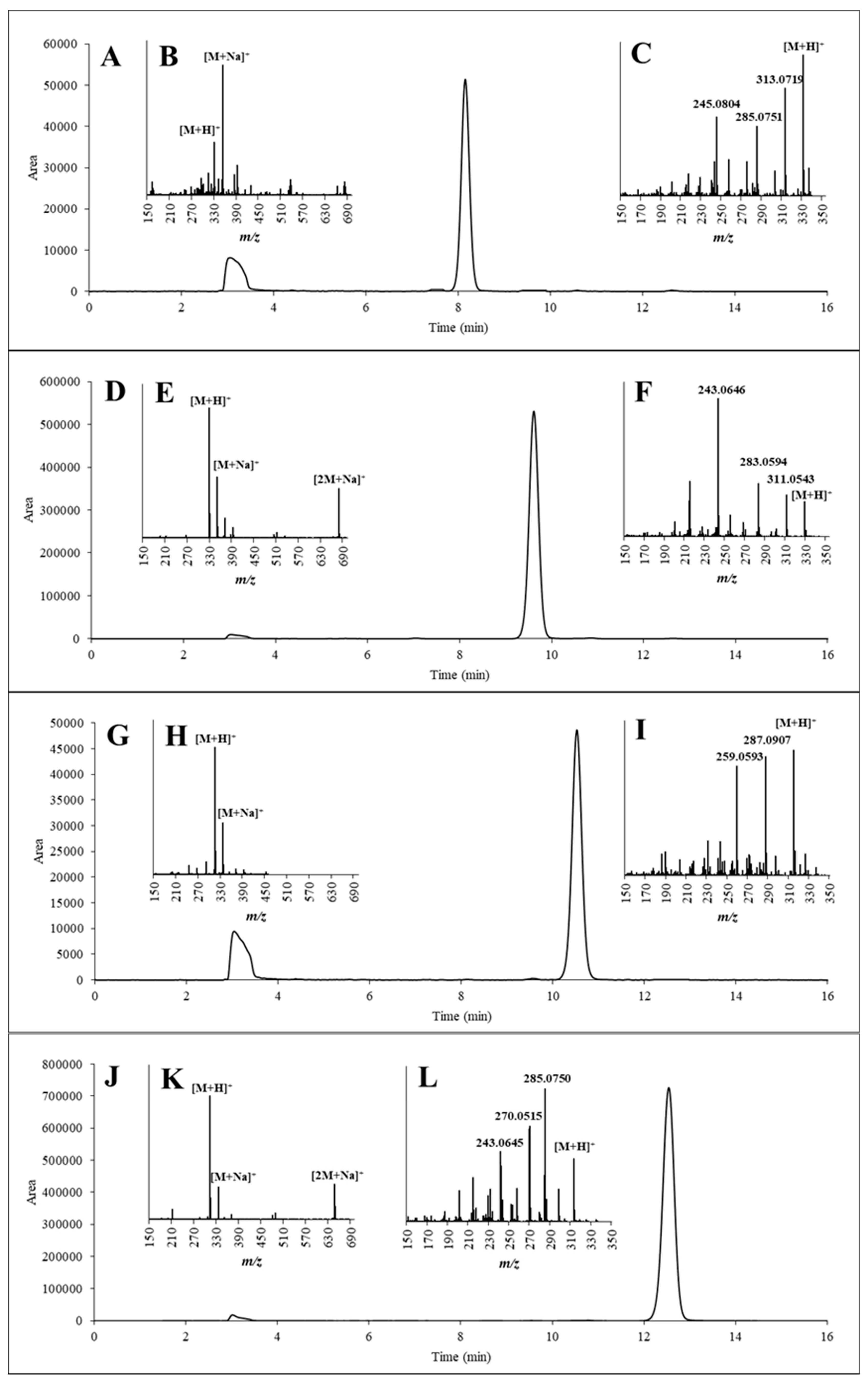
2.4. HPLC-UV and HR-MS to Verify Identity and Determine Purity
2.5. Yield of the Entire Purification Procedure
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Solvents
4.2. The Producer Strain and Culture Conditions
4.3. Aflatoxin Extraction
4.4. Testing of the Solvent Systems
4.5. HPLC-UV Measurements
4.6. Evaluation of the Separation
4.7. Centrifugal Partition Chromatography
4.8. HR-MS Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, D.L.; Gallagher, E.P. Mechanisms of aflatoxin carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 135–172. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Baranyi, N.; Chandrasekaran, M.; Vágvölgyi, Cs.; Kocsubé, S. Mycotoxin producers in the Aspergillus genus: An update. Acta Biol. Szeged. 2015, 59, 151–167. [Google Scholar]
- Nakai, V.K.; Rocha, L.O.; Goncalez, E.; Fonseca, H.; Ortega, E.M.M.; Corrêa, B. Distribution of fungi and aflatoxins in a stored peanut variety. Food Chem. 2008, 106, 285–290. [Google Scholar] [CrossRef]
- Rustom, I.Y.S. Aflatoxin in food and feed: Occurrence, legislation and inactivation by physical methods. Food Chem. 1997, 59, 57–67. [Google Scholar] [CrossRef]
- Calderari, T.O.; Iamanaka, B.T.; Frisvad, J.C.; Pitt, J.I.; Sartori, D.; Pereira, J.L.; Fungaro, M.H.; Taniwaki, M.H. The biodiversity of Aspergillus section Flavi in brazil nuts: From rainforest to consumer. Int. J. Food Microbiol. 2013, 160, 267–272. [Google Scholar] [CrossRef]
- Johnsson, P.; Linbald, M.; Thim, M.; Johnsson, N.; Vargas, E.A.; Medeiros, N.L.; Brabet, C.; Quaresma, M.; Olsen, M. Growht of aflatoxigenic moulds and aflatoxin formation in Brazil nuts. World Mycotoxin J. 2008, 1, 127–137. [Google Scholar] [CrossRef]
- Bakırdere, S.; Bora, S.; Bakırdere, E.G.; Aydın, F.; Arslan, Y.; Komesli, O.T.; Aydın, I.; Yıldırım, E. Aflatoxin species: Their health effects and determination methods in different foodstuffs. Cent. Eur. J. Chem. 2012, 10, 675–685. [Google Scholar] [CrossRef]
- Alexander, J.; Benford, D.; Cockburn, A.; Cravedi, J.P.; Dogliotti, E.; Dmenico, A.D. Effects on public health of an increase of the levels for aflatoxin total form 4 µg/kg to 10 µg/kg for tree nuts other than almonds, hazelnuts and pistachios. J. EFSA 2009, 1168, 1–11. [Google Scholar]
- Hepsag, F.; Golge, O.; Kabak, B. Quantitation of aflatoxins in pistachios and groundnuts using HPLC-FLD method. Food Control 2014, 38, 75–81. [Google Scholar] [CrossRef]
- Golge, O.; Hepsag, F.; Kabak, B. Determination of aflatoxins in walnut sujuk and Turkish delight by HPLC-FLD method. Food Control 2016, 59, 731–736. [Google Scholar] [CrossRef]
- Roma, A.D.; Rossini, C.; Riteni, A. A survey on the aflatoxin M1 occurrence and seasonal variation in buffalo and cow milk from southern Italy. Food Control 2017, 81, 30–33. [Google Scholar] [CrossRef]
- Herzallah, S.M. Determination of aflatoxins in eggs, milk, meat and meat products using HPLC fluorescent and UV detectors. Food Chem. 2009, 114, 1141–1146. [Google Scholar] [CrossRef]
- Campos, W.E.O.; Rosas, L.B.; Neto, A.P. Extended validation of a sensitive and robust method for simultaneous quantification of aflatoxins B1, B2, G1 and G2 in Brazil nuts by HPLC-FLD. J. Food Compos. Anal. 2017, 60, 90–96. [Google Scholar] [CrossRef]
- Kabak, B. Aflatoxins in hazelnuts and dried figs: Occurrence and exposure assessment. Food Chem. 2016, 211, 8–16. [Google Scholar] [CrossRef]
- Shiran, A.Y.; Tan, G.H.; Wong, R.C.S. Method validation in the determination of aflatoxins in noodle samples using the QuEChERS method (Quick, Easy, Cheap, Effective, Rugged and Safe) and high performance liquid chromatography coupled to a fluorescence detector (HPLC-FLD). Food Control 2011, 22, 1807–1813. [Google Scholar]
- Kong, W.; Wei, R.; Logrieco, A.F. Occurrence of toxigenic fungi and determination of mycotoxins by HPLC-FLD in functional foods and species in China markets. Food Chem. 2014, 146, 320–326. [Google Scholar] [CrossRef]
- Saldan, N.C.; Almeida, R.T.R.; Avíncola, A. Development for an analytical method for identification of Aspergillus flavus based on chemical markers using HPLC-MS. Food Chem. 2018, 241, 113–121. [Google Scholar] [CrossRef]
- Beltrán, E.; Ibáñez, M.; Sancho, J.V. UHPLC-MS/MS highly sensitive determination of aflatoxins, the aflatoxin metabolite M1 and ochratoxin A in baby food and milk. Food Chem. 2011, 126, 737–744. [Google Scholar] [CrossRef]
- Catharino, R.R.; Marques, L.A.; Santos, L.S. Aflatoxin screening my MALDI-TOF mass spectrometry. Anal. Chem. 2005, 77, 8155–8157. [Google Scholar] [CrossRef]
- Luo, X.; Wang, R.; Wang, L. Analyses by UPLC Q-TOF MS of products of aflatoxin B1 after ozone treatment. Food Addit. Contam. Part A 2014, 31, 105–110. [Google Scholar] [CrossRef]
- Mao, J.; Zheng, N.; Wen, F. Multi-mycotoxin analysis in raw milk by ultra high performance liquid chromatography coupled to quadrupole orbitrap mass spectrometry. Food Control 2018, 84, 305–311. [Google Scholar] [CrossRef]
- Martínez-Domínguez, G.; Romero-González, R.; Frenich, A.G. Multi-class methodology to determine pesticides and mycotoxins in green tea and royal jelly supplements by liquid chromatography coupled to Orbitrap high resolution mass spectrometry. Food Chem. 2016, 197, 907–915. [Google Scholar] [CrossRef]
- Reinholds, I.; Pugajeva, I.; Bartkevics, V. A reliable screening of mycotoxins and pesticide residues in paprika using ultra high performance liquid chromatography coupled to high resolution Orbitrap mass spectrometry. Food Control 2016, 60, 683–689. [Google Scholar] [CrossRef]
- Jia, W.; Chu, X.; Ling, Y. Multi-mycotoxin analysis in dairy products by liquid chromatography coupled to quadrupole orbitrap mass spectrometry. J. Chromatogr. A 2014, 1345, 107–114. [Google Scholar] [CrossRef]
- Sargenat, K.; Sheridan, A.; O’Kelly, J. Toxicity associated with certain samples of groundnuts. Nature 1961, 192, 1096–1097. [Google Scholar] [CrossRef]
- Nesbitt, B.F.; O’Kelly, J.; Sargeant, K. Aspergillus flavus and Turkey X disease: Toxic metabolites of Aspergillus flavus. Nature 1962, 195, 1062–1063. [Google Scholar] [CrossRef]
- Hartley, R.D.; Nesbitt, B.F.; O’Kelly, J. Toxic metabolites of Aspergillus flavus. Nature 1963, 198, 1056–1058. [Google Scholar] [CrossRef]
- Stubblefield, R.D.; Shotwell, O.L.; Shannon, G.M. Aflatoxins B1, B2, G1 and G2: Separation and purification. J. Am. Oil Chem. Soc. 1968, 45, 686–688. [Google Scholar] [CrossRef]
- De Jesus, A.E.; Gorst-Allman, C.P.; Horak, R.M. Large-scale purification of the mycotoxins aflatoxin B1, B2 and G1. J. Chromatogr. 1988, 450, 101–104. [Google Scholar] [CrossRef]
- Foucault, A.P. Centrifugal Partition Chromatography; Marcel Dekker: New York, NY, USA, 1995. [Google Scholar]
- Friesen, J.B.; Pauli, G.F. G.U.E.S.S.—A generally useful estimate of solvent systems for CCC. J. Liq. Chrom. Relat. Technol. 2005, 28, 2777–2806. [Google Scholar] [CrossRef]
- DeAmicis, C.; Edwards, N.A.; Giles, M.B. Comparison of preparative reversed phase liquid chromatography and countercurrent chromatography for the kilogram scale purification of crude spinetoram insecticide. J. Chromatogr. A 2011, 1218, 6122–6127. [Google Scholar] [CrossRef]
- Jeon, J.S.; Park, C.L.; Syed, A.S. Preparative separation of sesamin and sesamolin from defatted sesame meal via centrifugal partition chromatography with consecutive sample injection. J. Chromatogr. B 2016, 1011, 108–113. [Google Scholar] [CrossRef]
- Kotland, A.; Chollet, S.; Diard, C. Industrial case study on alkaloids purification by pH-zone refining centrifugal partition chromatography. J. Chromatogr. A 2016, 1474, 59–70. [Google Scholar] [CrossRef]
- Lee, J.H.; Ko, J.Y.; Oh, J.Y. Preparative isolation and purification of phlorotannins from Ecklonia cava using centrifugal partition chromatography by one-step. Food Chem. 2014, 158, 433–437. [Google Scholar] [CrossRef]
- Amarouche, N.; Boudesocque, L.; Sayagh, C. Purification of a modified cyclosporine A by co-current centrifugal partition chromatography: Process development and intensification. J. Chromatogr. A 2013, 1311, 72–78. [Google Scholar] [CrossRef]
- Bouju, E.; Berthold, A.; Faure, K. Carnosol purification. Scaling up centrifugal partition chromatography purifications. J. Chromatogr. A 2016, 1466, 59–66. [Google Scholar] [CrossRef]
- Szekeres, A.; Lorántfy, L.; Bencsik, O. Rapid purification method for fumonisin B1 using centrifugal partition chromatography. Food Addit. Contam. Part A 2013, 30, 147–155. [Google Scholar] [CrossRef]
- Hübner, F.; Harrer, H.; Fraske, A. Large scale purification of B-type fumonisins using centrifugal partition chromatography (CPC). Mycotoxin Res. 2012, 28, 37–43. [Google Scholar] [CrossRef]
- Onji, Y.; Aoki, Y.; Yamazoe, Y. Isolation of nivalenol and fusarenon-x from pressed barley culture by centrifugal partition chromatography. J. Liq. Chromatogr. 1988, 12, 2537–2546. [Google Scholar] [CrossRef]
- He, J.; Yang, R.; Zhou, T. Purification of deoxynivalenol from Fusarium graminearum rice culture and mouldy corn by high-speed countercurrent chromatography. J. Chromatogr. A 2007, 1151, 187–192. [Google Scholar] [CrossRef]
- He, J.; Tsao, R.; Yang, R. Purification of patulin from Penicillim expansum culture: High-speed counter-current chromatography (HSCCC) versus preparative high-performance liquid chromatography (prep-HPLC). Food Addit. Contam. Part A 2009, 29, 101–107. [Google Scholar] [CrossRef]
- Frisvad, J.C.; Hubka, V.; Ezekiel, C.N. Taxonomy of Aspergillus section Flavi and their production of aflatoxins, ochratoxins and other mycotoxins. Stud. Mycol. 2019, 93, 1–63. [Google Scholar] [CrossRef]
- Gömöri, Cs.; Nacsa-Farkas, E.; Kerekes, E.B. Effect of essential oil vapours on aflatoxin production of Aspergillus parasiticus. World Mycotoxin J. 2018, 11, 579–588. [Google Scholar] [CrossRef]
- Shih, C.N.; Marth, E.H. A procedure for rapid recovery of aflatoxins from cheese and other foods. J. Food Prot. 1971, 34, 119–123. [Google Scholar] [CrossRef]
- Foucault, A.P.; Chevolot, L. Counter-current chromatography: Instrumentation, solvent selection and some recent applications to natural product purification. J. Chromatogr. A 1998, 808, 3–22. [Google Scholar] [CrossRef]
- Garcia, M.; Blanco, J.L.; Suarez, G. Aflatoxins B1 and G1 solubility in standard solutions and stability during storage. Mycotoxin Res. 1994, 10, 97–100. [Google Scholar] [CrossRef]
- Beaver, R.W. Degradation of Aflatoxins in common HPLC solvents. J. High Res. Chromatogr. 1990, 13, 833–835. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent System | Volume Ratio | PImp1 | PImp2 | PAFG2 | PAFG1 | PAFB2 | PAFB1 | |
|---|---|---|---|---|---|---|---|---|
| 1 | hexane/chloroform/acetonitrile | 55:5.5:39.5 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 |
| 2 | 55:3.8:41.2 | 0.15 | 0.13 | 0.14 | 0.15 | 0.14 | 0.15 | |
| 3 | 77.7:3.2:19.1 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 4 | 77.7:5:17.3 | 0.19 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 5 | 42:9:49 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 6 | 34:8:58 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 7 | 55:7:38 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 8 | 63:10:27 | <0.10 | <0.10 | <0.10 | <0.10 | 0.11 | <0.10 | |
| 9 | 62:9:29 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 10 | hexane/acetone/water | 36:39:25 | 0.14 | <0.10 | <0.10 | <0.10 | 0.14 | <0.10 |
| 11 | 10:50:40 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 12 | 9:39:52 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 13 | 15:60:25 | 0.14 | 0.13 | 0.19 | 0.24 | 0.26 | 0.29 | |
| 14 | 56:24:20 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 15 | 66:24:10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 16 | 50:40:10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 17 | 30:60:10 | <0.10 | <0.10 | <0.10 | 0.11 | 0.13 | 0.15 | |
| 18 | 40:50:10 | 0.16 | 0.14 | 0.23 | 0.24 | 0.29 | 0.32 | |
| 19 | 32:63:5 | 0.13 | 0.13 | 0.19 | 0.21 | 0.24 | 0.26 | |
| 20 | 23:77:10 | 0.53 | 0.61 | 0.62 | 0.64 | 0.66 | 0.68 | |
| 21 | 20:70:10 | <0.10 | <0.10 | 0.14 | 0.14 | 0.18 | 0.20 | |
| 22 | heptane/acetone/water | 44:29:27 | 0.17 | 0.15 | 0.16 | 0.19 | 0.18 | 0.20 |
| 23 | 8:65:27 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 24 | 27:57:16 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 25 | 30:60:10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 26 | 40:20:40 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 27 | 20:10:70 a | - | - | - | - | - | - | |
| 28 | 55:25:20 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 29 | 80.5:9.5:10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 30 | 23:57:20 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 31 | 22:38:40 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | <0.10 | |
| 32 | toluene/acetone/water | 39:26:35 | 1.81 | 2.98 | 5.77 | 9.80 | 11.28 | 22.67 |
| 33 | 14:26:60 | 2.16 | 3.42 | 6.26 | 9.11 | 11.58 | 18.53 | |
| 34 | 10:40:50 | 8.33 | 38.64 | 6.46 | 4.11 | 2.66 | 58.49 | |
| 35 | 14:51:35 | 6.59 | 3.26 | 2.80 | 3.58 | 3.39 | 5.14 | |
| 36 | 29:55:16 | 1.39 | 1.83 | 2.86 | 3.86 | 5.00 | 5.03 | |
| 37 | 30:10:60 | 0.87 | 2.04 | 4.21 | 9.03 | 22.75 | 19.06 | |
| 38 | 6:74:20 b | - | - | - | - | - | - | |
| 39 | 18:12:70 | 0.86 | 1.72 | 2.27 | 6.16 | 13.11 | 12.32 | |
| 40 | 12:67:21 | 1.21 | 1.26 | 1.40 | 1.51 | 1.53 | 1.60 | |
| 41 | 17:67:16 | 1.08 | 1.11 | 1.16 | 1.21 | 1.25 | 1.25 | |
| 42 | 8:67:25 | 1.21 | 1.26 | 1.43 | 1.63 | 1.67 | 1.75 | |
| 43 | 70:10:20 | 0.99 | 1.78 | 2.90 | 5.38 | 9.92 | 9.42 | |
| 44 | 35:55:10 | 1.36 | 1.64 | 2.46 | 3.24 | 4.13 | 4.00 | |
| 45 | 15:55:30 | 1.38 | 1.58 | 2.19 | 2.83 | 2.84 | 3.36 | |
| 46 | 6:55:39 | 1.40 | 1.62 | 2.20 | 2.84 | 3.02 | 3.41 |
| Solvent System | Volume Ratio | PImp1 | PImp2 | PAFG2 | PAFG1 | PAFB2 | PAFB1 | |
|---|---|---|---|---|---|---|---|---|
| 1 | diethyl ether/acetic acid/water | 75:5:20 | 0.70 | 0.86 | 1.67 | 1.75 | 3.40 | 3.43 |
| 2 | 60:20:20 | 0.26 | 0.32 | 0.46 | 0.53 | 0.66 | 4.00 | |
| 3 | 30:10:60 | 0.10 | 0.11 | 0.21 | 0.22 | 0.25 | 0.43 | |
| 4 | 45:15:40 | <0.10 | 0.15 | 0.19 | 0.20 | 0.33 | 0.41 | |
| 5 | chloroform/acetic acid/water | 26:24:50 | 0.42 | 0.37 | <0.10 | <0.10 | <0.10 | <0.10 |
| 6 | 36:34:30 | 0.47 | 0.93 | 0.18 | 0.16 | 0.11 | <0.10 | |
| 7 | 35:45:20 | 1.03 | 1.15 | 0.58 | 0.57 | 0.29 | 0.84 | |
| 8 | toluene/acetic acid/water | 80:6:14 | <0.10 | <0.10 | 0.31 | 0.62 | 1.56 | 1.88 |
| 9 | 20:10:70 | 0.14 | 0.43 | 0.73 | 1.70 | 3.18 | 4.57 | |
| 10 | 30:10:60 | <0.10 | 0.4 | 0.63 | 1.36 | 1.73 | 3.33 | |
| 11 | 20:20:60 | <0.10 | 0.3 | 0.3 | 0.63 | 0.94 | 2.65 | |
| 12 | 30:24:50 | 0.04 | 0.14 | 0.18 | 0.36 | 0.54 | 1.21 | |
| 13 | 20:30:50 | <0.10 | 0.12 | 0.16 | 0.28 | 0.56 | 0.92 | |
| 14 | 40:30:30 | <0.10 | <0.10 | <0.10 | 0.16 | 0.42 | 0.47 | |
| 15 | 63:30:7 | <0.10 | <0.10 | <0.10 | 0.10 | 0.23 | 0.20 | |
| 16 | 40:42:18 | <0.10 | 0.11 | 0.11 | <0.10 | 0.23 | 0.22 | |
| 17 | 20:55:25 | <0.10 | <0.10 | <0.10 | <0.10 | 0.31 | 0.21 |
| AFB1 | AFB2 | AFG1 | AFG2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c Ref. (m/z) | d Mass dev. (ppm) | e Ratio dev. (%) | c Ref. (m/z) | d Mass dev. (ppm) | e Ratio dev. (%) | c Ref. (m/z) | d Mass dev. (ppm) | e Ratio dev. (%) | c Ref. (m/z) | d Mass dev. (ppm) | e Ratio dev. (%) | |
| a Full scan | 313.0699 | 1.25 | 0 | 315.0849 | 0.99 | 0 | 329.0646 | 1.43 | 0 | 331.0811 | 1.17 | 0 |
| 335.0504 | 1.56 | 0 | 337.0669 | 1.18 | 0 | 351.0464 | 1.26 | 0 | 353.0631 | 1.66 | 0 | |
| 647.1142 | 2.02 | 0 | - | - | - | 679.1039 | 2.54 | 0 | - | - | - | |
| b MS2 | 285.0750 | 1.99 | 5 | 287.0907 | 1.37 | 1 | 311.0543 | 2.57 | 1 | 313.0719 | 2.03 | 0 |
| 270.0515 | 1.04 | 3 | 259.0593 | 1.18 | 2 | 283.0594 | 1.79 | 3 | 285.0751 | 1.98 | 1 | |
| 243.0645 | 2.30 | 1 | - | - | - | 243.0646 | 2.01 | 4 | 245.0804 | 2.13 | 1 | |
| AFB1 | AFB2 | AFG1 | AFG2 | Total AFs | ||
|---|---|---|---|---|---|---|
| Crude extract (culture medium → dichloromethane) | Yield (mg) | 442 | 40 | 827 | 105 | 1414 |
| Purity (%) | 39 | 4.0 | 45.4 | 2.9 | 91.3 | |
| Second extract (dichloromethane → hexane/methanol/water) | Yield (mg) | 442 | 40 | 827 | 105 | 1414 |
| Purity (%) | 41.7 | 4.0 | 46.9 | 3.0 | 95.6 | |
| Recovery (%) | 100 | 100 | 100 | 100 | 100 | |
| Third extract (hexane/methanol/water → dichloromethane) | Yield (mg) | 442 | 40 | 827 | 105 | 14141 |
| Purity (%) | 42.6 | 4.2 | 47.5 | 3.0 | 97.3 | |
| Recovery (%) | 100 | 100 | 100 | 100 | 100 | |
| CPC separation (final product) (30:24:50 toluene/acetic acid/water) | Yield (mg) | 400 | 34 | 817 | 100 | 1351 |
| Purity (%) | 98.2 | 96.3 | 98.1 | 97.0 | 97.3 | |
| Recovery (%) | 90.5 | 85.3 | 98.7 | 96.0 | 92.6 | |
| Whole procedure | Recovery (%) | 90.5 | 85.3 | 98.7 | 96.0 | 92.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endre, G.; Hegedüs, Z.; Turbat, A.; Škrbić, B.; Vágvölgyi, C.; Szekeres, A. Separation and Purification of Aflatoxins by Centrifugal Partition Chromatography. Toxins 2019, 11, 309. https://doi.org/10.3390/toxins11060309
Endre G, Hegedüs Z, Turbat A, Škrbić B, Vágvölgyi C, Szekeres A. Separation and Purification of Aflatoxins by Centrifugal Partition Chromatography. Toxins. 2019; 11(6):309. https://doi.org/10.3390/toxins11060309
Chicago/Turabian StyleEndre, Gábor, Zsófia Hegedüs, Adiyadolgor Turbat, Biljana Škrbić, Csaba Vágvölgyi, and András Szekeres. 2019. "Separation and Purification of Aflatoxins by Centrifugal Partition Chromatography" Toxins 11, no. 6: 309. https://doi.org/10.3390/toxins11060309
APA StyleEndre, G., Hegedüs, Z., Turbat, A., Škrbić, B., Vágvölgyi, C., & Szekeres, A. (2019). Separation and Purification of Aflatoxins by Centrifugal Partition Chromatography. Toxins, 11(6), 309. https://doi.org/10.3390/toxins11060309