Chemical Synthesis, Proper Folding, Nav Channel Selectivity Profile and Analgesic Properties of the Spider Peptide Phlotoxin 1


 , ,
, , 
Abstract
:1. Introduction
2. Results
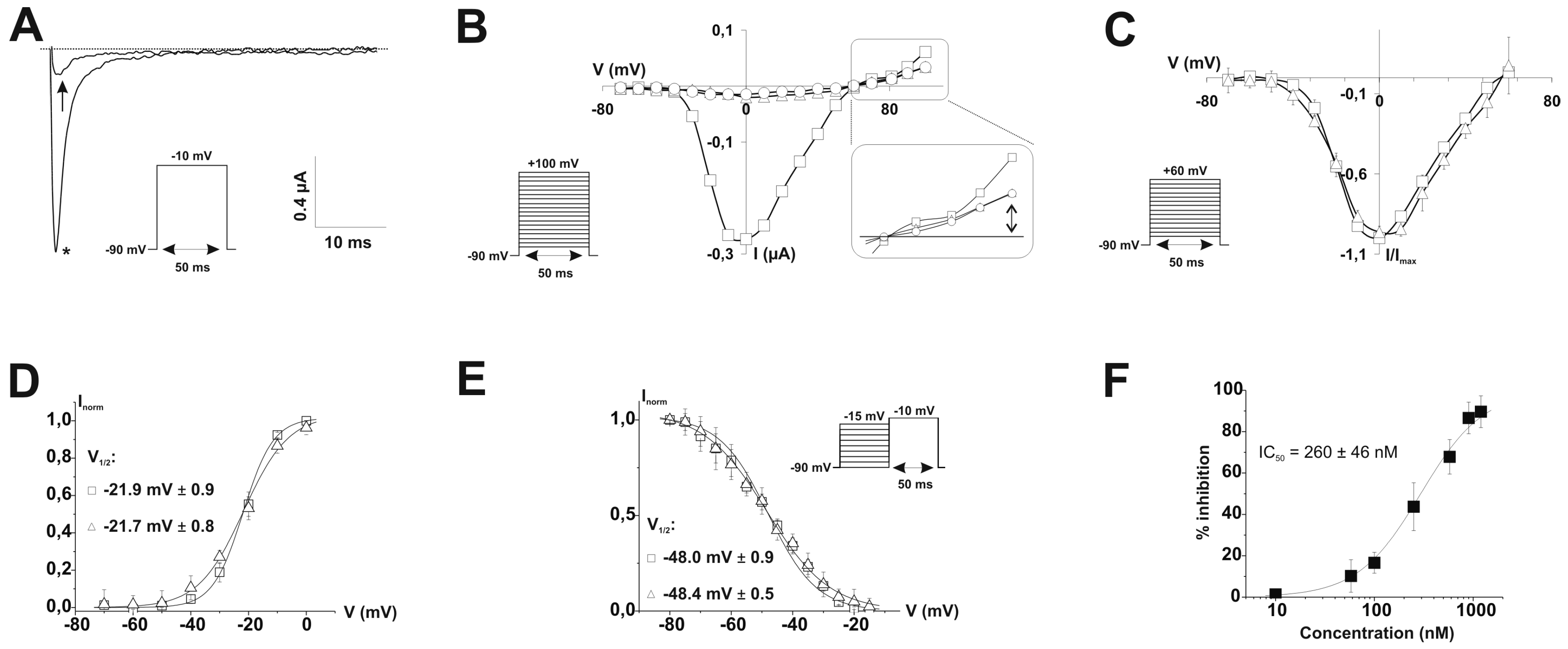
2.1. PhlTx1 Description
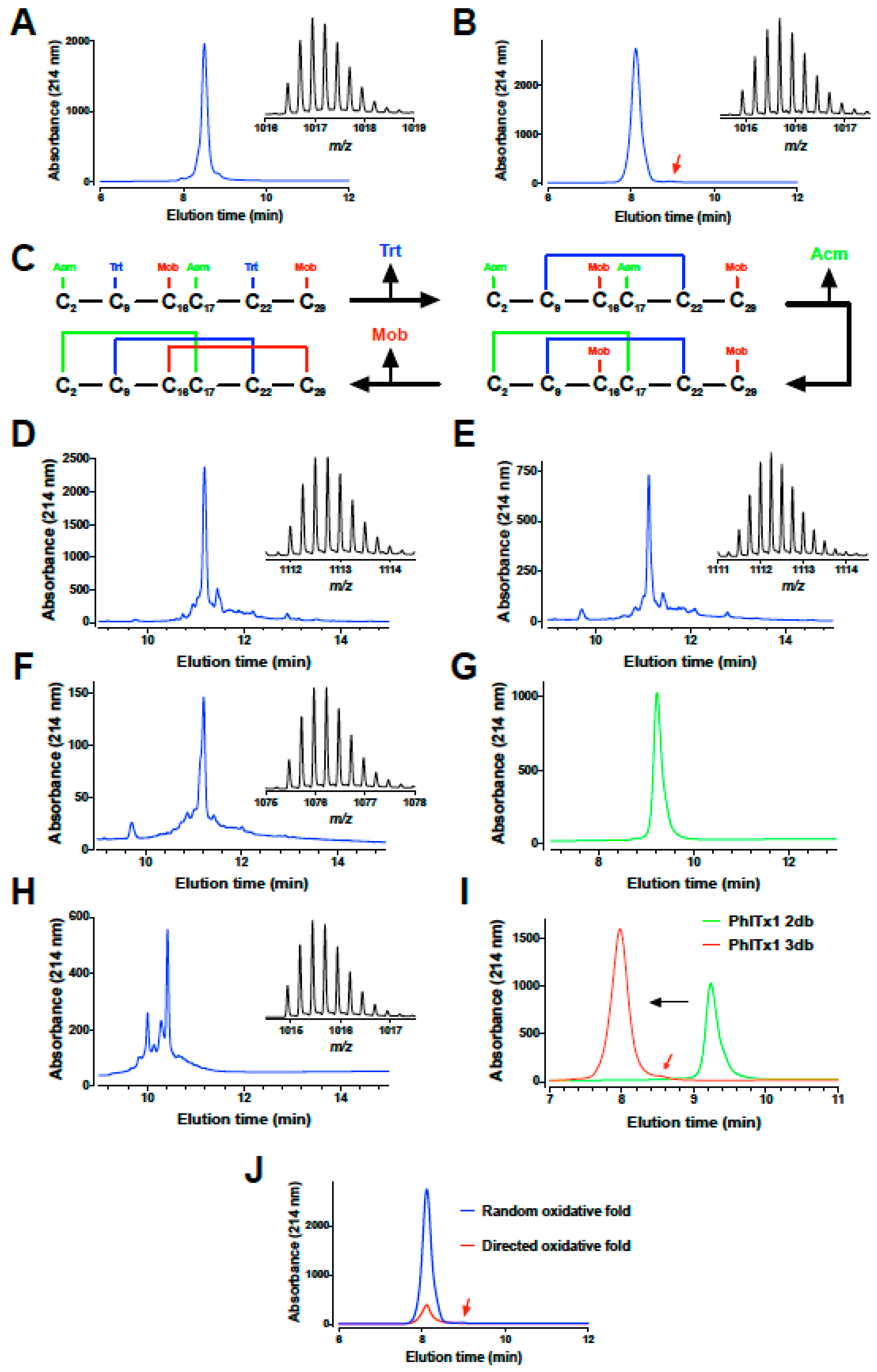
2.2. PhlTx1 Chemical Synthesis
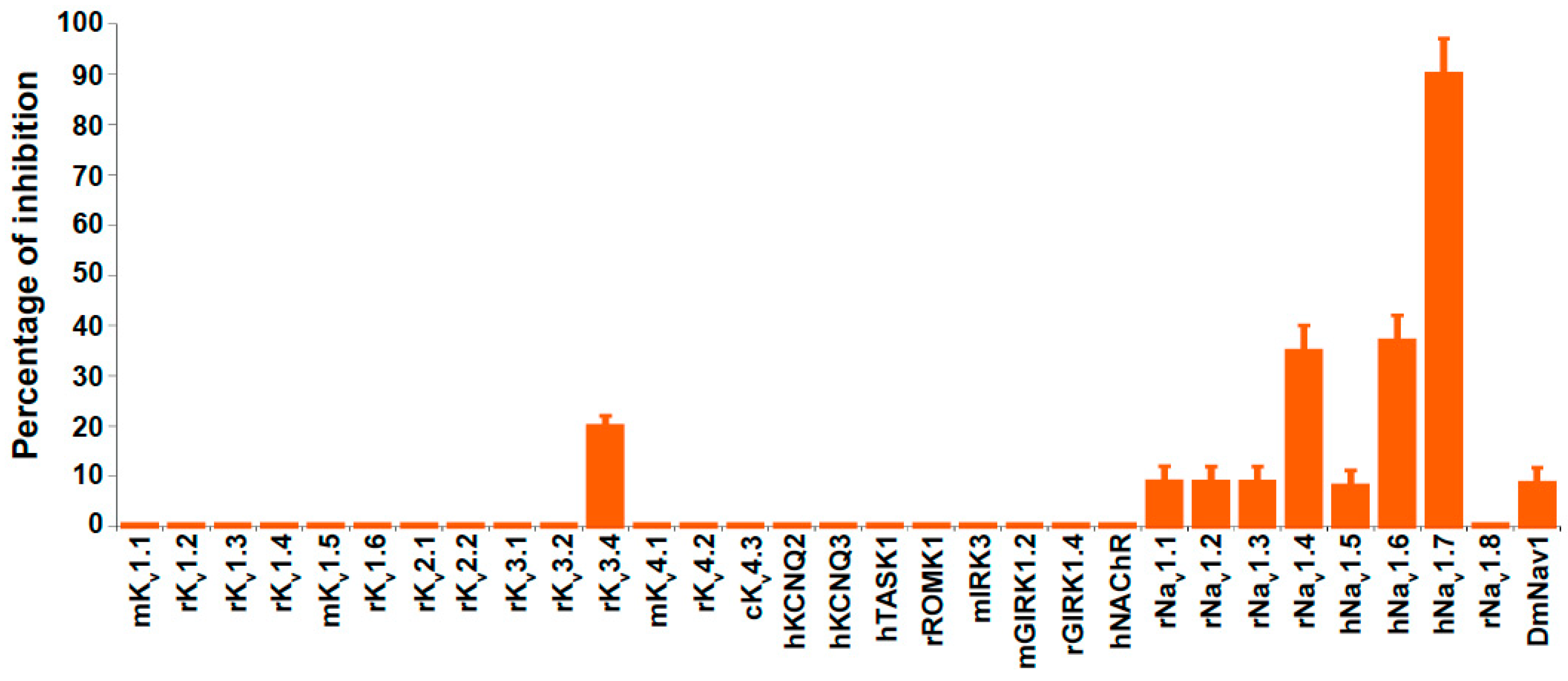
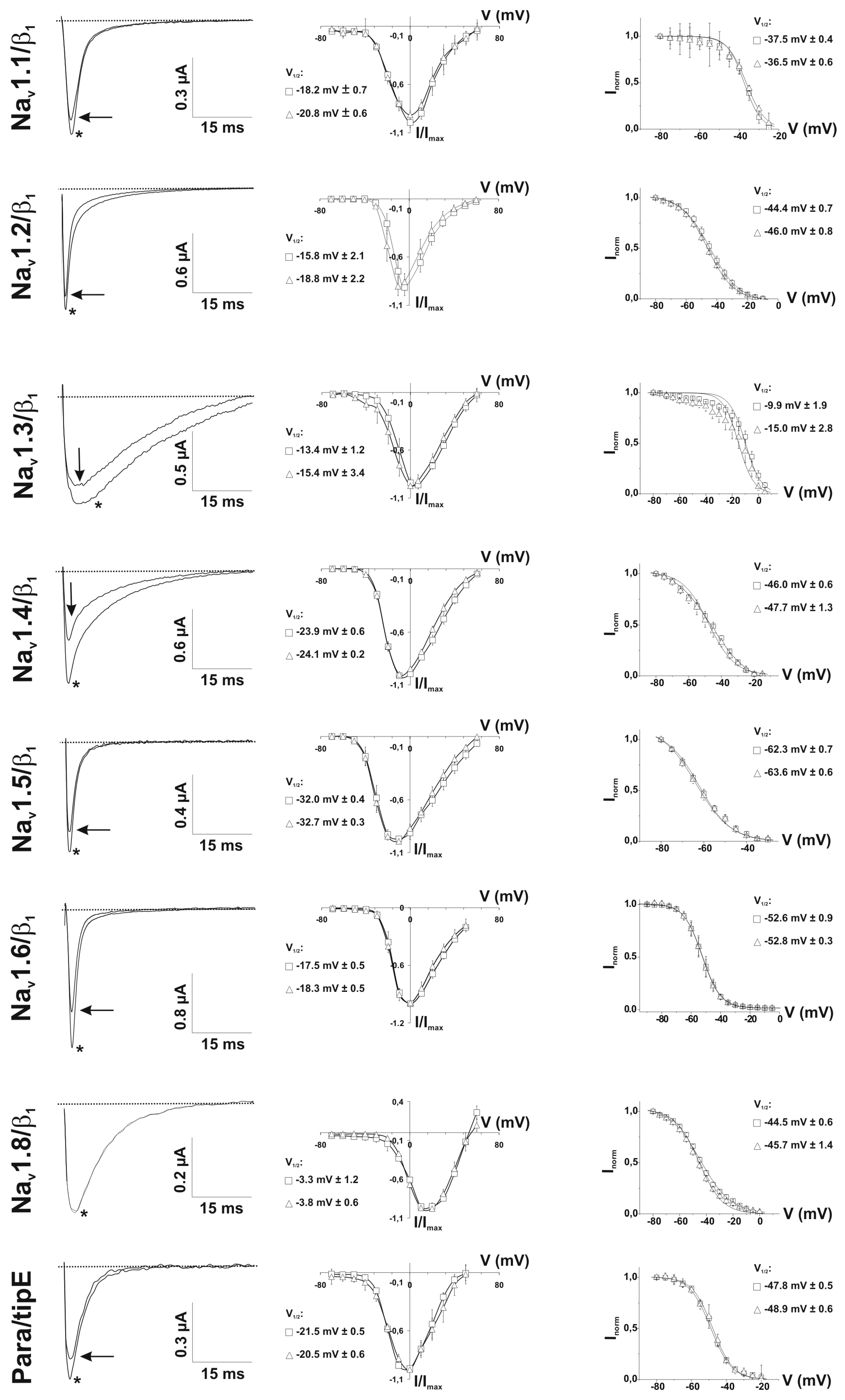
2.3. Ion Channel Selectivity of PhlTx1 and Preferential Activity on Nav1.7
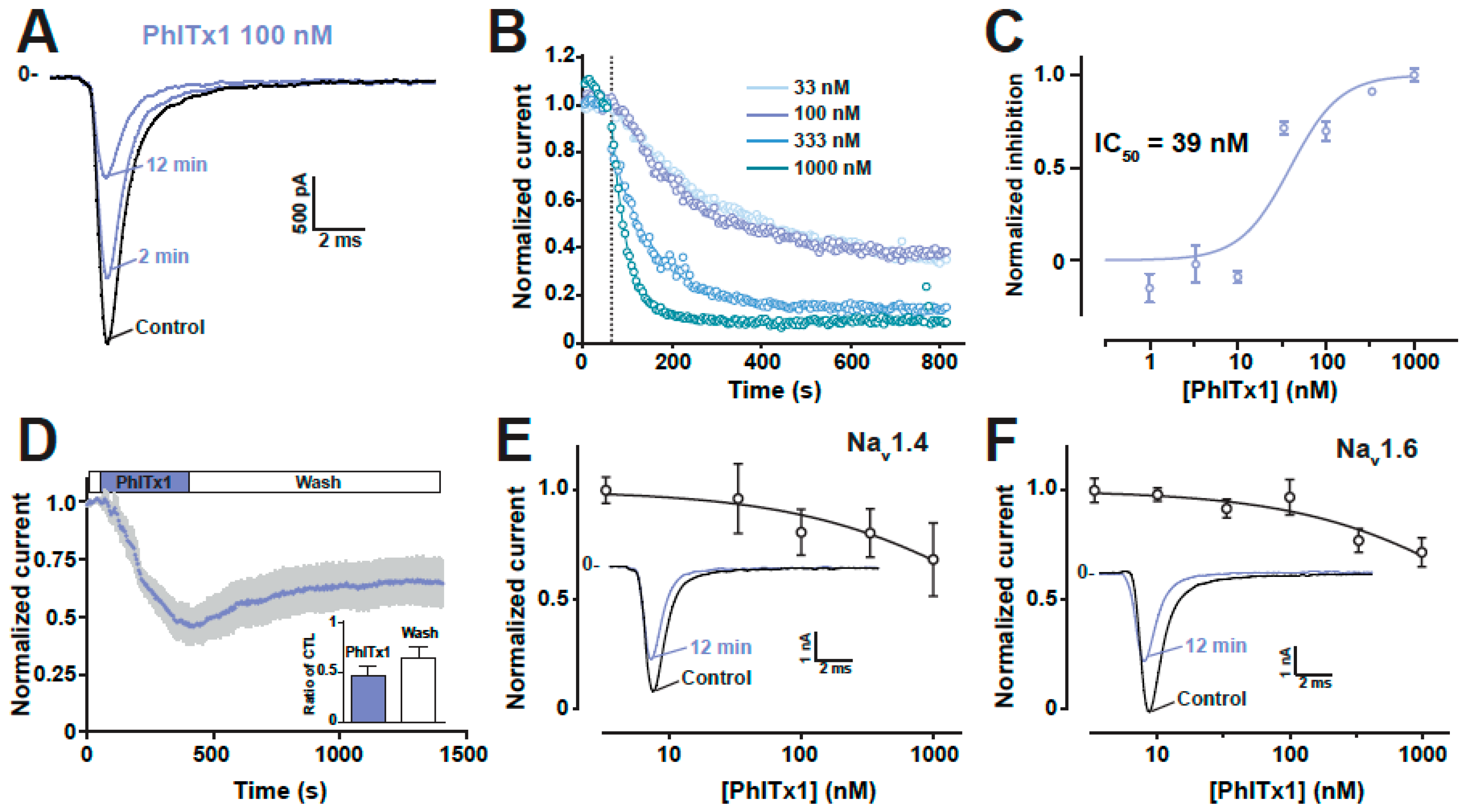
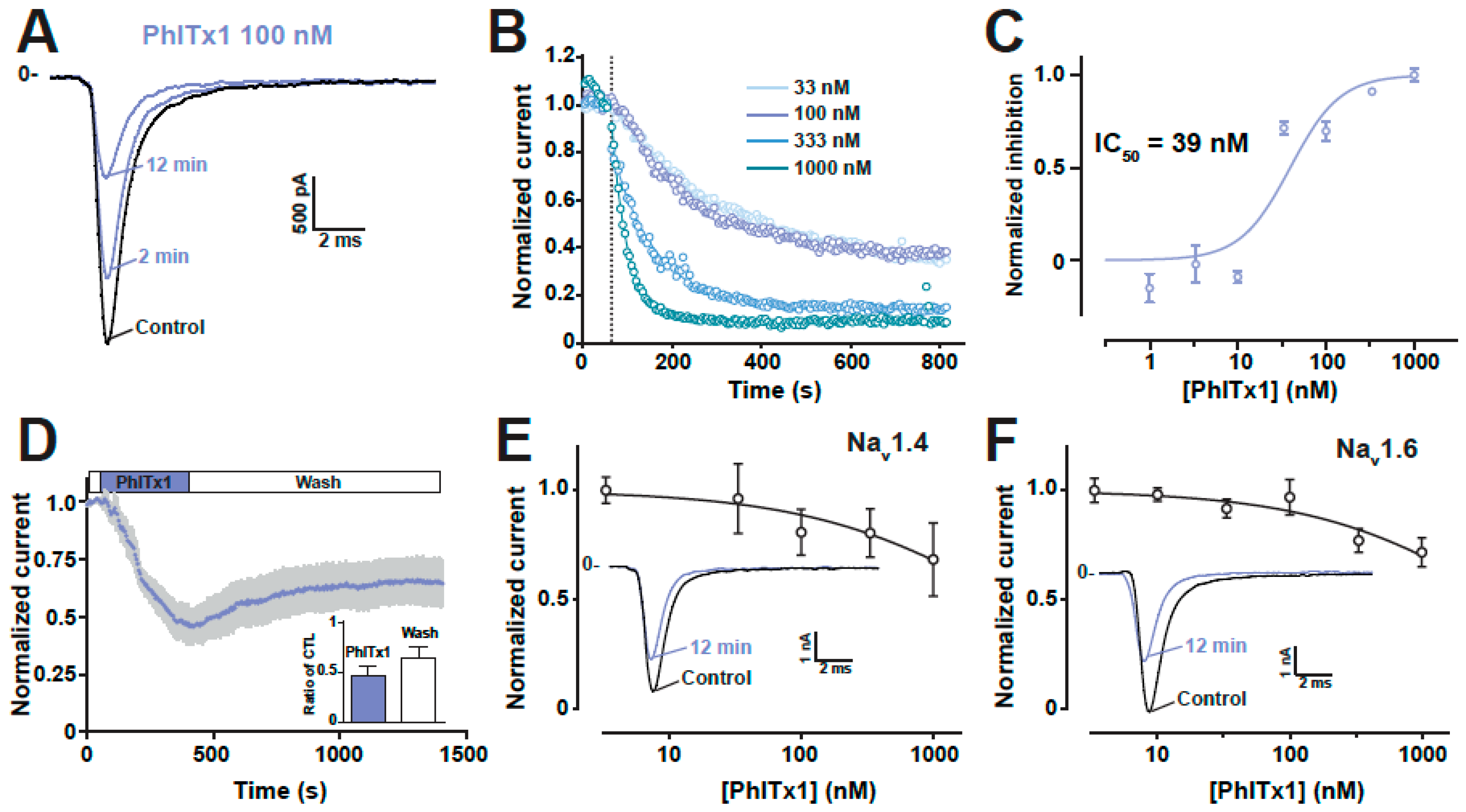
2.4. Refined Affinity of PhlTx1 for the Nav1.7 Channel in Mammalian Cells
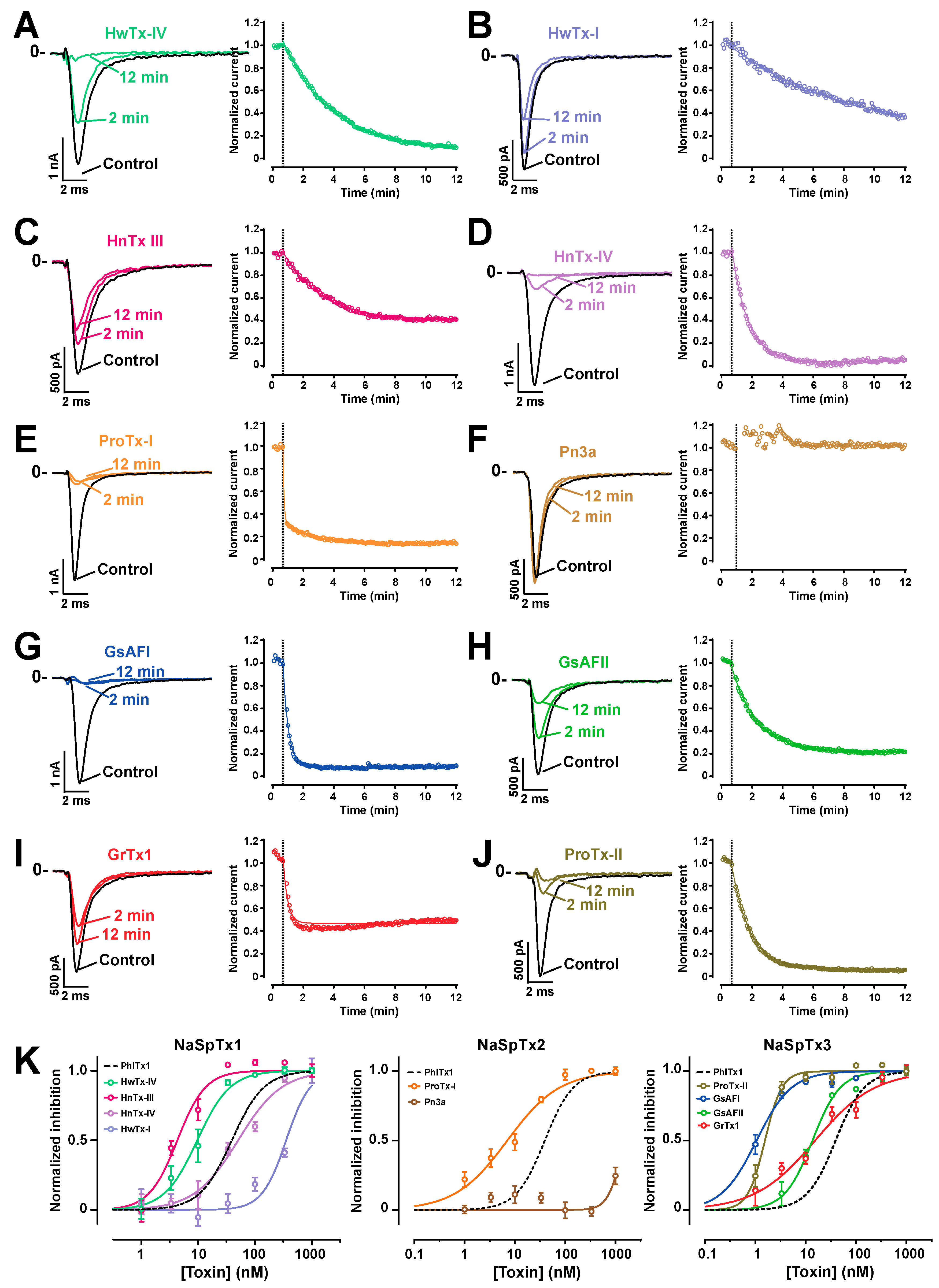
2.5. Comparison of the Nav1.7 Channel Blocking Efficience of PhlTx1 with That of Leading Toxins Active on Nav1.7
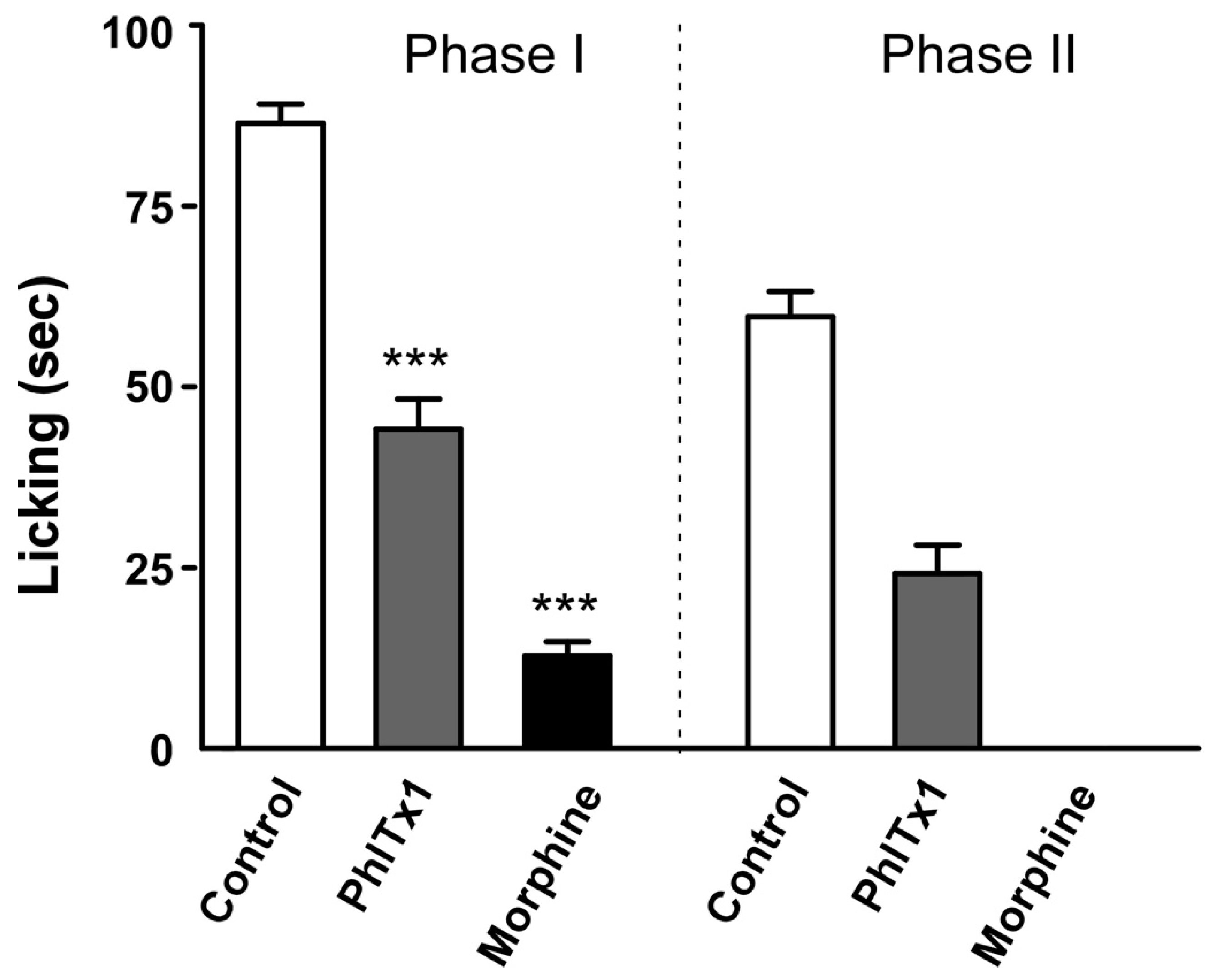
2.6. Analgesic Potential of PhlTx1 In Vivo
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Peptides
5.2. Chemical Synthesis of PhlTx1
5.3. Cell Culture
5.4. Xenopus Oocyte Expression and Recording Experiments
5.5. Pharmacological Applications Using the Automated Patch-Clamp System
5.6. Formalin Pain Test
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Lauria, G.; Merkies, I.S.; Cheng, X.; Han, C.; Ahn, H.S.; Persson, A.K.; Hoeijmakers, J.G.; Gerrits, M.M.; Pierro, T.; et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 19444–19449. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates Inc.: Sunderland, MA, USA, 2001; Volume 1. [Google Scholar]
- Leipold, E.; Liebmann, L.; Korenke, G.C.; Heinrich, T.; Giesselmann, S.; Baets, J.; Ebbinghaus, M.; Goral, R.O.; Stodberg, T.; Hennings, J.C.; et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet. 2013, 45, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Ahern, C.A.; Payandeh, J.; Bosmans, F.; Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 2016, 147, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Bregman, H.; Berry, L.; Buchanan, J.L.; Chen, A.; Du, B.; Feric, E.; Hierl, M.; Huang, L.; Immke, D.; Janosky, B.; et al. Identification of a potent, state-dependent inhibitor of Nav1.7 with oral efficacy in the formalin model of persistent pain. J. Med. Chem. 2011, 54, 4427–4445. [Google Scholar] [CrossRef] [PubMed]
- Chakka, N.; Bregman, H.; Du, B.; Nguyen, H.N.; Buchanan, J.L.; Feric, E.; Ligutti, J.; Liu, D.; McDermott, J.S.; Zou, A.; et al. Discovery and hit-to-lead optimization of pyrrolopyrimidines as potent, state-dependent Na(v)1.7 antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Focken, T.; Liu, S.; Chahal, N.; Dauphinais, M.; Grimwood, M.E.; Chowdhury, S.; Hemeon, I.; Bichler, P.; Bogucki, D.; Waldbrook, M.; et al. Discovery of Aryl Sulfonamides as Isoform-Selective Inhibitors of NaV1.7 with Efficacy in Rodent Pain Models. ACS Med. Chem. Lett. 2016, 7, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.M.; DeGoey, D.A.; Shi, L.; Gum, R.J.; Fricano, M.M.; Lundgaard, G.L.; El-Kouhen, O.F.; Hsieh, G.C.; Neelands, T.; Matulenko, M.A.; et al. Substituted Indazoles as Nav1.7 Blockers for the Treatment of Pain. J. Med. Chem. 2016, 59, 3373–3391. [Google Scholar] [CrossRef]
- Graceffa, R.F.; Boezio, A.A.; Able, J.; Altmann, S.; Berry, L.M.; Boezio, C.; Butler, J.R.; Chu-Moyer, M.; Cooke, M.; DiMauro, E.F.; et al. Sulfonamides as Selective NaV1.7 Inhibitors: Optimizing Potency, Pharmacokinetics, and Metabolic Properties to Obtain Atropisomeric Quinolinone (AM-0466) that Affords Robust in Vivo Activity. J. Med. Chem. 2017, 60, 5990–6017. [Google Scholar] [CrossRef]
- Ho, G.D.; Tulshian, D.; Bercovici, A.; Tan, Z.; Hanisak, J.; Brumfield, S.; Matasi, J.; Heap, C.R.; Earley, W.G.; Courneya, B.; et al. Discovery of pyrrolo-benzo-1,4-diazines as potent Na(v)1.7 sodium channel blockers. Bioorg. Med. Chem. Lett. 2014, 24, 4110–4113. [Google Scholar] [CrossRef]
- Macsari, I.; Besidski, Y.; Csjernyik, G.; Nilsson, L.I.; Sandberg, L.; Yngve, U.; Ahlin, K.; Bueters, T.; Eriksson, A.B.; Lund, P.E.; et al. 3-Oxoisoindoline-1-carboxamides: Potent, state-dependent blockers of voltage-gated sodium channel Na(V)1.7 with efficacy in rat pain models. J. Med. Chem. 2012, 55, 6866–6880. [Google Scholar] [CrossRef] [PubMed]
- Marx, I.E.; Dineen, T.A.; Able, J.; Bode, C.; Bregman, H.; Chu-Moyer, M.; DiMauro, E.F.; Du, B.; Foti, R.S.; Fremeau, R.T., Jr.; et al. Sulfonamides as Selective NaV1.7 Inhibitors: Optimizing Potency and Pharmacokinetics to Enable in Vivo Target Engagement. ACS Med. Chem. Lett. 2016, 7, 1062–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.N.; Bregman, H.; Buchanan, J.L.; Du, B.; Feric, E.; Huang, L.; Li, X.; Ligutti, J.; Liu, D.; Malmberg, A.B.; et al. Discovery and optimization of aminopyrimidinones as potent and state-dependent Nav1.7 antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Roecker, A.J.; Egbertson, M.; Jones, K.L.G.; Gomez, R.; Kraus, R.L.; Li, Y.; Koser, A.J.; Urban, M.O.; Klein, R.; Clements, M.; et al. Discovery of selective, orally bioavailable, N-linked arylsulfonamide Nav1.7 inhibitors with pain efficacy in mice. Bioorg. Med. Chem. Lett. 2017, 27, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jia, Q.; Zenova, A.Y.; Chafeev, M.; Zhang, Z.; Lin, S.; Kwan, R.; Grimwood, M.E.; Chowdhury, S.; Young, C.; et al. The discovery of benzenesulfonamide-based potent and selective inhibitors of voltage-gated sodium channel Na(v)1.7. Bioorg. Med. Chem. Lett. 2014, 24, 4397–4401. [Google Scholar] [CrossRef]
- Suzuki, S.; Kuroda, T.; Kimoto, H.; Domon, Y.; Kubota, K.; Kitano, Y.; Yokoyama, T.; Shimizugawa, A.; Sugita, R.; Koishi, R.; et al. Discovery of (phenoxy-2-hydroxypropyl)piperidines as a novel class of voltage-gated sodium channel 1.7 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5419–5423. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, Z.; Yang, G.; Teng, M.; Qin, J.; Hu, Z.; Hou, L.; Shen, L.; Dong, H.; Zhang, Y.; et al. The discovery of tetrahydropyridine analogs as hNav1.7 selective inhibitors for analgesia. Bioorg. Med. Chem. Lett. 2017, 27, 2210–2215. [Google Scholar] [CrossRef]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef]
- Gingras, J.; Smith, S.; Matson, D.J.; Johnson, D.; Nye, K.; Couture, L.; Feric, E.; Yin, R.; Moyer, B.D.; Peterson, M.L.; et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS ONE 2014, 9, e105895. [Google Scholar] [CrossRef]
- Fertleman, C.R.; Baker, M.D.; Parker, K.A.; Moffatt, S.; Elmslie, F.V.; Abrahamsen, B.; Ostman, J.; Klugbauer, N.; Wood, J.N.; Gardiner, R.M.; et al. SCN9A mutations in paroxysmal extreme pain disorder: Allelic variants underlie distinct channel defects and phenotypes. Neuron 2006, 52, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.G.; Han, C.; Merkies, I.S.; Macala, L.J.; Lauria, G.; Gerrits, M.M.; Dib-Hajj, S.D.; Faber, C.G.; Waxman, S.G. Small nerve fibres, small hands and small feet: A new syndrome of pain, dysautonomia and acromesomelia in a kindred with a novel NaV1.7 mutation. Brain 2012, 135, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.L.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef] [PubMed]
- Faber, C.G.; Hoeijmakers, J.G.; Ahn, H.S.; Cheng, X.; Han, C.; Choi, J.S.; Estacion, M.; Lauria, G.; Vanhoutte, E.K.; Gerrits, M.M.; et al. Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 2012, 71, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Dib-Hajj, S.D.; Waxman, S.G. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 2004, 24, 8232–8236. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128, 1847–1854. [Google Scholar] [CrossRef]
- Fischer, T.Z.; Waxman, S.G. Familial pain syndromes from mutations of the NaV1.7 sodium channel. Ann. N. Y. Acad. Sci. 2010, 1184, 196–207. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, C.; Wang, Z.; Chen, Y.; Xie, H.; Li, S.; Liu, X.; Liu, Z.; Chen, P. Mechanistic insights into Nav1.7-dependent regulation of rat prostate cancer cell invasiveness revealed by toxin probes and proteomic analysis. FEBS J. 2019. [Google Scholar] [CrossRef]
- Yildirim, S.; Altun, S.; Gumushan, H.; Patel, A.; Djamgoz, M.B. Voltage-gated sodium channel activity promotes prostate cancer metastasis in vivo. Cancer Lett. 2012, 323, 58–61. [Google Scholar] [CrossRef]
- Kis-Toth, K.; Hajdu, P.; Bacskai, I.; Szilagyi, O.; Papp, F.; Szanto, A.; Posta, E.; Gogolak, P.; Panyi, G.; Rajnavolgyi, E. Voltage-gated sodium channel Nav1.7 maintains the membrane potential and regulates the activation and chemokine-induced migration of a monocyte-derived dendritic cell subset. J. Immunol. 2011, 187, 1273–1280. [Google Scholar] [CrossRef]
- Muroi, Y.; Ru, F.; Kollarik, M.; Canning, B.J.; Hughes, S.A.; Walsh, S.; Sigg, M.; Carr, M.J.; Undem, B.J. Selective silencing of Na(V)1.7 decreases excitability and conduction in vagal sensory neurons. J. Physiol. 2011, 589, 5663–5676. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Pyrski, M.; Jacobi, E.; Bufe, B.; Willnecker, V.; Schick, B.; Zizzari, P.; Gossage, S.J.; Greer, C.A.; Leinders-Zufall, T.; et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature 2011, 472, 186–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, Y.P.; Price, N.; Namdari, R.; Cohen, C.J.; Lamers, M.H.; Winters, C.; Price, J.; Young, C.E.; Verschoof, H.; Sherrington, R.; et al. Treatment of Na(v)1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain 2012, 153, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, S.B.; London, C.; Abbadie, C.; Felix, J.P.; Garcia, M.L.; Jochnowitz, N.; Karanam, B.V.; Li, X.; Lyons, K.A.; McGowan, E.; et al. A novel benzazepinone sodium channel blocker with oral efficacy in a rat model of neuropathic pain. Bioorg. Med. Chem. Lett. 2013, 23, 3640–3645. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S.; Mukund, S.; Deng, L.; Khakh, K.; Chang, E.; Ho, H.; Shriver, S.; Young, C.; Lin, S.; Johnson, J.P., Jr.; et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 2015, 350, aac5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosmans, F.; Swartz, K.J. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [Green Version]
- Gilchrist, J.; Bosmans, F. Animal toxins can alter the function of Nav1.8 and Nav1.9. Toxins 2012, 4, 620–632. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Schmalhofer, W.A.; Calhoun, J.; Burrows, R.; Bailey, T.; Kohler, M.G.; Weinglass, A.B.; Kaczorowski, G.J.; Garcia, M.L.; Koltzenburg, M.; Priest, B.T. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 2008, 74, 1476–1484. [Google Scholar] [CrossRef] [PubMed]
- Maertens, C.; Cuypers, E.; Amininasab, M.; Jalali, A.; Vatanpour, H.; Tytgat, J. Potent modulation of the voltage-gated sodium channel Nav1.7 by OD1, a toxin from the scorpion Odonthobuthus doriae. Mol. Pharmacol. 2006, 70, 405–414. [Google Scholar] [CrossRef]
- Yang, S.; Xiao, Y.; Kang, D.; Liu, J.; Li, Y.; Undheim, E.A.; Klint, J.K.; Rong, M.; Lai, R.; King, G.F. Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc. Natl. Acad. Sci. USA 2013, 110, 17534–17539. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Dekan, Z.; Wingerd, J.S.; Smith, J.J.; Munasinghe, N.R.; Bhola, R.F.; Imlach, W.L.; Herzig, V.; Armstrong, D.A.; Rosengren, K.J.; et al. Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci. Rep. 2017, 7, 40883. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, T.C.; Benoit, E.; Partiseti, M.; Servent, D. The NaV1.7 Channel Subtype as an Antinociceptive Target for Spider Toxins in Adult Dorsal Root Ganglia Neurons. Front. Pharmacol. 2018, 9, 1000. [Google Scholar] [CrossRef]
- Bosmans, F.; Escoubas, P.; Diochot, S.; Mebs, D.; Craik, D.; Hill, J.; Nakajima, T.; Lazdunski, M.; Tytgat, J. Isolation and characterization of phlotoxin 1 (PhlTx1), a novel peptide active on voltage-gated sodium channels. In Proceedings of the 13ème Rencontres en Toxinologie “Toxines et douleur”, Paris, France, 1–2 December 2005. [Google Scholar]
- Emery, E.C.; Luiz, A.P.; Wood, J.N. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin. Ther. Targets 2016, 20, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Blacklow, B.; Kornhauser, R.; Hains, P.G.; Loiacono, R.; Escoubas, P.; Graudins, A.; Nicholson, G.M. alpha-Elapitoxin-Aa2a, a long-chain snake alpha-neurotoxin with potent actions on muscle (alpha1)(2)betagammadelta nicotinic receptors, lacks the classical high affinity for neuronal alpha7 nicotinic receptors. Biochem. Pharmacol. 2011, 81, 314–325. [Google Scholar] [CrossRef]
- Xiao, Y.; Bingham, J.P.; Zhu, W.; Moczydlowski, E.; Liang, S.; Cummins, T.R. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J. Biol. Chem. 2008, 283, 27300–27313. [Google Scholar] [CrossRef]
- Liu, Z.; Cai, T.; Zhu, Q.; Deng, M.; Li, J.; Zhou, X.; Zhang, F.; Li, D.; Li, J.; Liu, Y.; et al. Structure and function of hainantoxin-III, a selective antagonist of neuronal tetrodotoxin-sensitive voltage-gated sodium channels isolated from the Chinese bird spider Ornithoctonus hainana. J. Biol. Chem. 2013, 288, 20392–20403. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, J.; Zhang, Y.; Xun, X.; Tang, D.; Peng, D.; Yi, J.; Liu, Z.; Shi, X. Synthesis and analgesic effects of mu-TRTX-Hhn1b on models of inflammatory and neuropathic pain. Toxins 2014, 6, 2363–2378. [Google Scholar] [CrossRef]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven novel modulators of the analgesic target NaV 1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [CrossRef]
- Redaelli, E.; Cassulini, R.R.; Silva, D.F.; Clement, H.; Schiavon, E.; Zamudio, F.Z.; Odell, G.; Arcangeli, A.; Clare, J.J.; Alagon, A.; et al. Target promiscuity and heterogeneous effects of tarantula venom peptides affecting Na+ and K+ ion channels. J. Biol. Chem. 2010, 285, 4130–4142. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Cai, T.F.; Li, W.Y.; Zhang, H.; Liu, Y.B.; Peng, K.; Liang, S.; Zhang, D.Y. Functional expression of spider neurotoxic peptide huwentoxin-I in E. coli. PLoS ONE 2011, 6, e21608. [Google Scholar] [CrossRef]
- Middleton, R.E.; Warren, V.A.; Kraus, R.L.; Hwang, J.C.; Liu, C.J.; Dai, G.; Brochu, R.M.; Kohler, M.G.; Gao, Y.D.; Garsky, V.M.; et al. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 2002, 41, 14734–14747. [Google Scholar] [CrossRef]
- Park, J.H.; Carlin, K.P.; Wu, G.; Ilyin, V.I.; Musza, L.L.; Blake, P.R.; Kyle, D.J. Studies examining the relationship between the chemical structure of protoxin II and its activity on voltage gated sodium channels. J. Med. Chem. 2014, 57, 6623–6631. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Martin-Eauclaire, M.F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, V.; Pennington, M.W.; Norton, R.S.; Tarcha, E.J.; Londono, L.M.; Sims-Fahey, B.; Upadhyay, S.K.; Lakey, J.T.; Iadonato, S.; Wulff, H.; et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012, 59, 529–546. [Google Scholar] [CrossRef] [Green Version]
- Craik, D.J.; Adams, D.J. Chemical modification of conotoxins to improve stability and activity. ACS Chem. Biol. 2007, 2, 457–468. [Google Scholar] [CrossRef]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef]
- Altafaj, X.; Joux, N.; Ronjat, M.; De Waard, M. Oocyte expression with injection of purified T7 RNA polymerase. Methods Mol. Biol. 2006, 322, 55–67. [Google Scholar] [CrossRef]
- Geib, S.; Sandoz, G.; Carlier, E.; Cornet, V.; Cheynet-Sauvion, V.; De Waard, M. A novel Xenopus oocyte expression system based on cytoplasmic coinjection of T7-driven plasmids and purified T7-RNA polymerase. Recept. Channels 2001, 7, 331–343. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | NaSpTx1 | NaSpTx2 | NaSpTx3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Toxin | PhlTx1 | HnTx-III | HnTx-IV | HwTx-I | HwTx-IV | ProTx-I | Pn3a | GrTx1 | ProTx-II | GsAFI | GsAFII |
| IC50 | 39 | 50.6 | 4.3 | 25.1 | 9.6 | 7.1 | 1457 | 15.3 | 1.5 | 1.0 | 13.6 |
| Publ. | 250 | 232 | 21 | 630 | 26 | 72 | 0.9 | 370 | 1 | 40 | 1030 |
| Ref. | [47] | [51] | [52] | [55] | [50] | [56] | [45] | [54] | [57] | [54] | [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolas, S.; Zoukimian, C.; Bosmans, F.; Montnach, J.; Diochot, S.; Cuypers, E.; De Waard, S.; Béroud, R.; Mebs, D.; Craik, D.; et al. Chemical Synthesis, Proper Folding, Nav Channel Selectivity Profile and Analgesic Properties of the Spider Peptide Phlotoxin 1. Toxins 2019, 11, 367. https://doi.org/10.3390/toxins11060367
Nicolas S, Zoukimian C, Bosmans F, Montnach J, Diochot S, Cuypers E, De Waard S, Béroud R, Mebs D, Craik D, et al. Chemical Synthesis, Proper Folding, Nav Channel Selectivity Profile and Analgesic Properties of the Spider Peptide Phlotoxin 1. Toxins. 2019; 11(6):367. https://doi.org/10.3390/toxins11060367
Chicago/Turabian StyleNicolas, Sébastien, Claude Zoukimian, Frank Bosmans, Jérôme Montnach, Sylvie Diochot, Eva Cuypers, Stephan De Waard, Rémy Béroud, Dietrich Mebs, David Craik, and et al. 2019. "Chemical Synthesis, Proper Folding, Nav Channel Selectivity Profile and Analgesic Properties of the Spider Peptide Phlotoxin 1" Toxins 11, no. 6: 367. https://doi.org/10.3390/toxins11060367
APA StyleNicolas, S., Zoukimian, C., Bosmans, F., Montnach, J., Diochot, S., Cuypers, E., De Waard, S., Béroud, R., Mebs, D., Craik, D., Boturyn, D., Lazdunski, M., Tytgat, J., & De Waard, M. (2019). Chemical Synthesis, Proper Folding, Nav Channel Selectivity Profile and Analgesic Properties of the Spider Peptide Phlotoxin 1. Toxins, 11(6), 367. https://doi.org/10.3390/toxins11060367