Simultaneous Detection of 14 Microcystin Congeners from Tissue Samples Using UPLC- ESI-MS/MS and Two Different Deuterated Synthetic Microcystins as Internal Standards


 ,
,
Abstract
:1. Introduction
2. Results
2.1. Method Establishment and Optimisation
2.2. Use of Internal Standard
2.3. Method Validation
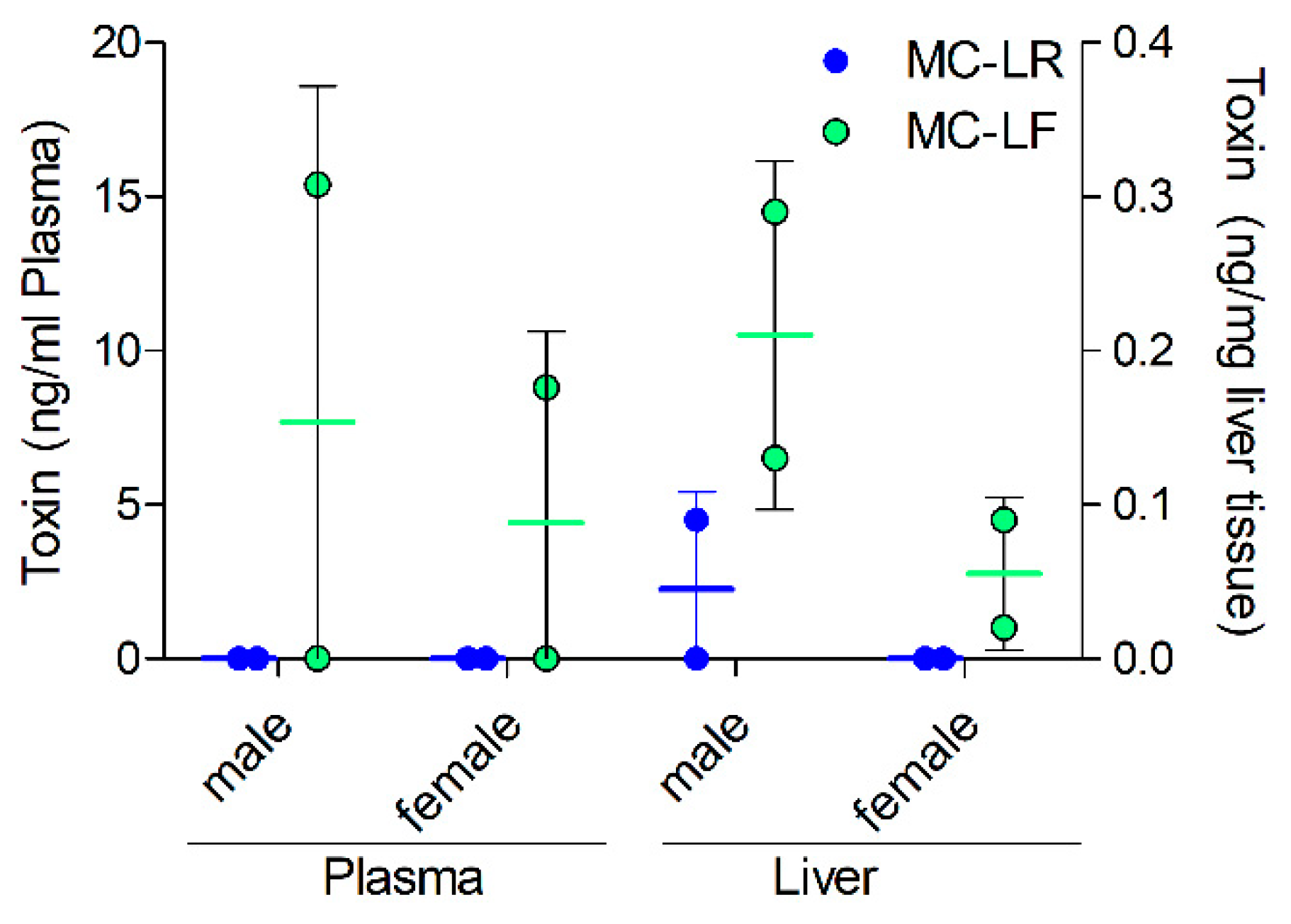
2.4. MC Levels in Exposed Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sample Generation for the Establishment and Validation
4.3. Extraction Method for MC from Blood and Liver Tissue Samples
4.4. UPLC-MS/MS Analysis
4.5. Animal Samples
4.6. Data Analyses and Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, S.M.; Carmichael, W.W.; Jochimsen, E.M.; Rinehart, K.L.; Lau, S.; Shaw, G.R.; Eaglesham, G.K. Human intoxication by microcystins during renal dialysis treatment in Caruaru—Brazil. Toxicology 2002, 181–182, 441–446. [Google Scholar] [CrossRef]
- Spoof, L.; Catherine, A. Appendix 3: Tables of Microcystins and Nodularin. In Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis; Meriluoto, J., Spoof, L., Codd, G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Fischer, W.J.; Altheimer, S.; Cattori, V.; Meier, P.J.; Dietrich, D.R.; Hagenbuch, B. Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol. Appl. Pharmacol. 2005, 203, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Hoeger, S.J.; Stemmer, K.; Feurstein, D.J.; Knobeloch, D.; Nussler, A.; Dietrich, D.R. The role of organic anion transporting polypeptides (OATPs/SLCOs) in the toxicity of different microcystin congeners in vitro: A comparison of primary human hepatocytes and OATP-transfected HEK293 cells. Toxicol. Appl. Pharmacol. 2010, 245, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Dietrich, D.R. The role of human MRP2 in cellular efflux of microcystin congeners (Poster). In Proceedings of the 57th Annual Meeting of the Society of Toxicology, San Antonio, TX, USA, 11–15 March 2018. [Google Scholar]
- Kaur, G.; Zemskov, I.; Wittmann, V.; Dietrich, D.R. Toxicokinetics and dynamics of microcystin congeners are structure-dependent (Poster). In Proceedings of the 58th Annual Meeting of the Society of Toxicology, Baltimore, MA, USA, 10–14 March 2018. [Google Scholar]
- Kondo, F.; Ikai, Y.; Oka, H.; Okumura, M.; Ishikawa, N.; Harada, K.; Matsuura, K.; Murata, H.; Suzuki, M. Formation, characterization, and toxicity of the glutathione and cysteine conjugates of toxic heptapeptide microcystins. Chem. Res. Toxicol. 1992, 5, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Swingle, M.; Ni, L.; Honkanen, R.E. Small Molecule Inhibitors of ser/thr protein phosphatases: Specificity, use and common forms of abuse. Methods Mol. Bio 2007, 365, 23–38. [Google Scholar] [CrossRef]
- Garibo, D.; Flores, C.; Ceto, X.; Prieto-Simon, B.; Del Valle, M.; Caixach, J.; Diogene, J.; Campas, M. Inhibition equivalency factors for microcystin variants in recombinant and wild-type protein phosphatase 1 and 2A assays. Environ. Sci. Pollut. Res. Int. 2014, 18, 10652–10660. [Google Scholar] [CrossRef] [PubMed]
- Mattila, K.; Annila, A.; Rantala, T.T. Metals ions mediate the binding of cyanobacterial toxins to human protein phosphatase 1—A computational study. Acta Universitatis Ouluensis A 2000, A 351, 31–68. [Google Scholar]
- Fontanillo, M.; Köhn, M. Microcystins: Synthesis and structure–activity relationship studies toward PP1 and PP2A. Bioorg. Med. Chem. 2018, 26, 1118–1126. [Google Scholar] [CrossRef]
- Sanseverino, I.; António, D.C.; Loos, R.; Lettieri, T. Cyanotoxins: Methods and Approaches for Their Analysis and Detection; Centre, J.R., Ed.; Publications Office of the European Union: Luxembourg, 2017. [Google Scholar]
- Hawkins, P.R.; Novic, S.; Cox, P.; Neilan, B.A.; Burns, B.P.; Shaw, G.; Wickramasinghe, W.; Peerapornpisal, Y.; Ruangyuttikarn, W.; Itayama, T. A review of analytical methods for assessing the public health risk from microcystin in the aquatic environment. J. Water Supply Res. Technol. 2005, 54, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Gilroy, D.J.; Kauffman, K.W.; Hall, R.A.; Huang, X.; Chu, F.S. Assessing potential health risks from microcystin toxins in blue-green algae dietary supplements. Environ. Health Perspect. 2000, 108, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, T. Microcystin Congeners Described in the Literature, Version 5. Available online: http://10.6084/m9.figshare.880756 (accessed on 2 May 2016).
- Sano, T.; Nohara, K.; Shiraishi, F.; Kaya, K. A Method for Micro-Determination of Total Microcystin Content in Waterblooms of Cyanobacteria (Blue-Green Algae). Int. J. Environ. Anal. Chem. 1992, 49, 163–170. [Google Scholar] [CrossRef]
- Neffling, M.-R.; Lance, E.; Meriluoto, J. Detection of free and covalently bound microcystins in animal tissues by liquid chromatography–tandem mass spectrometry. Environ. Pollut. 2010, 158, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Greer, B.; Maul, R.; Campbell, K.; Elliott, C.T. Detection of freshwater cyanotoxins and measurement of masked microcystins in tilapia from Southeast Asian aquaculture farms. Anal. Bioanal. Chem. 2017, 409, 4057–4069. [Google Scholar] [CrossRef] [PubMed]
- Mallet, C. Analysis of Microcystins RR, LY, and YR in Bottled, Tap, and Surface Water Using ACQUITY UPLC Systems with 2D-LC Technology. Available online: http://www.waters.com/waters/library.htm?cid=511436&lid=134826892&locale=de_DE (accessed on 9 November 2018).
- Turner, A.D.; Waack, J.; Lewis, A.; Edwards, C.; Lawton, L. Development and single-laboratory validation of a UHPLC-MS/MS method for quantitation of microcystins and nodularin in natural water, cyanobacteria, shellfish and algal supplement tablet powders. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1074–1075, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Manubolu, M.; Lee, J.; Riedl, K.M.; Kua, Z.X.; Collart, L.P.; Ludsin, S.A. Optimization of extraction methods for quantification of microcystin-LR and microcystin-RR in fish, vegetable, and soil matrices using UPLC-MS/MS. Harmful Algae 2018, 76, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Greer, B.; McNamee, S.E.; Boots, B.; Cimarelli, L.; Guillebault, D.; Helmi, K.; Marcheggiani, S.; Panaiotov, S.; Breitenbach, U.; Akcaalan, R.; et al. A validated UPLC-MS/MS method for the surveillance of ten aquatic biotoxins in European brackish and freshwater systems. Harmful Algae 2016, 55, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Diez-Quijada, L.; Guzman-Guillen, R.; Prieto Ortega, A.I.; Llana-Ruiz-Cabello, M.; Campos, A.; Vasconcelos, V.; Jos, A.; Camean, A.M. New Method for Simultaneous Determination of Microcystins and Cylindrospermopsin in Vegetable Matrices by SPE-UPLC-MS/MS. Toxins 2018, 10, 406. [Google Scholar] [CrossRef]
- Zervou, S.K.; Christophoridis, C.; Kaloudis, T.; Triantis, T.M.; Hiskia, A. New SPE-LC-MS/MS method for simultaneous determination of multi-class cyanobacterial and algal toxins. J. Hazard. Mater. 2017, 323, 56–66. [Google Scholar] [CrossRef]
- Roegner, A.F.; Schirmer, M.P.; Puschner, B.; Brena, B.; Gonzalez-Sapienza, G. Rapid Quantitative Analysis of Microcystins in Raw Surface Waters with MALDI MS Utilizing Easily Synthesized Internal Standards. Toxicon 2014, 78, 94–102. [Google Scholar] [CrossRef]
- Smith, J.L.; Boyer, G.L. Standardization of microcystin extraction from fish tissues: A novel internal standard as a surrogate for polar and non-polar variants. Toxicon 2009, 53, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Puddick, J.; Prinsep, M.R.; Wood, S.A.; Kaufononga, S.A.; Cary, S.C.; Hamilton, D.P. High levels of structural diversity observed in microcystins from Microcystis CAWBG11 and characterization of six new microcystin congeners. Mar. Drugs 2014, 12, 5372–5395. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.D.; Dhanji-Rapkova, M.; O’Neill, A.; Coates, L.; Lewis, A.; Lewis, K. Analysis of Microcystins in Cyanobacterial Blooms from Freshwater Bodies in England. Toxins 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, D.; Stemmer, K.; Kleinteich, J.; Speicher, T.; Dietrich, D.R. Microcystin Congener– and Concentration-Dependent Induction of Murine Neuron Apoptosis and Neurite Degeneration. Toxicol. Sci. 2011, 124, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Heussner, A.H.; Altaner, S.; Kamp, L.; Rubio, F.; Dietrich, D.R. Pitfalls in microcystin extraction and recovery from human blood serum. Chem. Biol. Interact. 2014, 223, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Altaner, S.; Puddick, J.; Wood, S.A.; Dietrich, D.R. Adsorption of Ten Microcystin Congeners to Common Laboratory-Ware Is Solvent and Surface Dependent. Toxins 2017, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline on Bioanalytical Method Validation; Committee for Medicinal Products for Human Use (CHMP), Ed.; European Medicines Agency: London, UK, 2011.
- Stokvis, E.; Rosing, H.; Beijnen, J.H. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: Necessity or not? Rapid Commun. Mass Spectrom. 2005, 19, 401–407. [Google Scholar] [CrossRef]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef]
- Carmichael, W.W.; Azevedo, S.; An, J.S.; Molica, R.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef]
- Feurstein, D.; Holst, K.; Fischer, A.; Dietrich, D.R. Oatp-associated uptake and toxicity of microcystins in primary murine whole brain cells. Toxicol. Appl. Pharmacol. 2009, 234, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Choudhuri, S.; Ogura, K.; Csanaky, I.L.; Lei, X.; Cheng, X.; Song, P.Z.; Klaassen, C.D. Characterization of organic anion transporting polypeptide 1b2-null mice: Essential role in hepatic uptake/toxicity of phalloidin and microcystin-LR. Toxicol. Sci. 2008, 103, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zemskov, I.; Kropp, H.M.; Wittmann, V. Regioselective Cleavage of Thioether Linkages in Microcystin Conjugates. Chem. Eur. J. 2016, 22, 10990–10997. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.O. Rapid and Convenient Oxidative Release of Thiol-Conjugated Forms of Microcystins for Chemical Analysis. Chem. Res. Toxicol. 2017, 30, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.O.; Sandvik, M.; Nonga, H.E.; Ballot, A.; Wilkins, A.L.; Rise, F.; Jaabaek, J.A.; Loader, J.I. Conjugation of Microcystins with Thiols is Reversible: Base-Catalyzed Deconjugation for Chemical Analysis. Chem. Res. Toxicol. 2016, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An overview of matrix effects in liquid chromatography–mass spectrometry. Mass Spectrom. Rev. 2011, 30, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.A.; Pace, J.G.; Matson, C.F.; Miura, G.A.; Lawrence, W.B. Tissue distribution, excretion and hepatic biotransformation of microcystin-LR in mice. J. Pharmacol. Exp. Ther. 1991, 256, 176–182. [Google Scholar]
- Cheng, X.; Maher, J.M.; Lu, H.; Klaassen, C.D. Endocrine regulation of gender-divergent mouse organic anion transporting polypeptide (Oatp) expression. Mol. Pharmacol. 2006, 70, 1291–1297. [Google Scholar] [CrossRef]
- Chen, J.; Xie, P.; Li, L.; Xu, J. First identification of the hepatotoxic microcystins in the serum of a chronically exposed human population together with indication of hepatocellular damage. Toxicol. Sci. 2009, 108, 81–89. [Google Scholar] [CrossRef]
- Zheng, C.; Zeng, H.; Lin, H.; Wang, J.; Feng, X.; Qiu, Z.; Chen, J.A.; Luo, J.; Luo, Y.; Huang, Y.; et al. Serum microcystin levels positively linked with risk of hepatocellular carcinoma: A case-control study in southwest China. Hepatology 2017, 66, 1519–1528. [Google Scholar] [CrossRef]
- Pflugmacher, S.; Wiegand, C.; Oberemm, A.; Beattie, K.A.; Krause, E.; Codd, G.A.; Steinberg, C.E.W. Identification of an enzymatically formed glutathione conjugate of the cyanobacterial hepatotoxin microcystin-LR: The first step of detoxication. Biochim. Biophys. Acta 1998, 1425, 527–533. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, P.; Chen, J.; Liang, G. Distribution of microcystins in various organs (heart, liver, intestine, gonad, brain, kidney and lung) of Wistar rat via intravenous injection. Toxicon 2008, 52, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, J.; Chen, J.; Xie, P. Excretion pattern and dynamics of glutathione detoxification of microcystins in Sprague Dawley rat. Chemosphere 2018, 191, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Greer, B.; Meneely, J.P.; Elliott, C.T. Uptake and accumulation of Microcystin-LR based on exposure through drinking water: An animal model assessing the human health risk. Sci. Rep. 2018, 8, 4913. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chen, M.; Anderson, J.T.; Sun, X.; Hu, S.; Sparreboom, A.; Baker, S.D. Interaction Between Sex and Organic Anion-Transporting Polypeptide 1b2 on the Pharmacokinetics of Regorafenib and Its Metabolites Regorafenib-N-Oxide and Regorafenib-Glucuronide in Mice. Clin. Transl. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zemskov, I.; Altaner, S.; Dietrich, D.R.; Wittmann, V. Total Synthesis of Microcystin-LF and Derivatives Thereof. J. Org. Chem. 2017, 82, 3680–3691. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solid Phase Extraction (SPE) | Liquid-Liquid Partitioning (LLP) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| StrataX | HLB | PRiME HLB | % in MeOH | % in n-Hexane | ||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| MC-RR | 63.4 | 2.3 | 90.4 | 6.0 | 100.9 | 5.8 | 89.0 | 9.9 | 0.6 | 0.0 |
| MC-YR | 112.7 | 2.2 | 81.2 | 5.6 | 80.6 | 9.8 | 96.4 | 4.6 | 1.0 | 0.1 |
| MC-LR | 79.5 | 2.5 | 95.1 | 5.3 | 97.8 | 7.4 | 98.5 | 4.8 | 0.5 | 0.0 |
| MC-FR | 66.8 | 12.4 | 97.6 | 6.9 | 107.2 | 7.2 | 97.8 | 7.0 | 0.6 | 0.0 |
| MC-WR | 74.9 | 2.2 | 98.5 | 6.6 | 114.7 | 7.9 | 97.8 | 8.5 | 0.5 | 0.0 |
| MC-RA | 87.7 | 5.5 | 104.8 | 6.6 | 98.0 | 4.3 | 97.3 | 4.2 | 0.6 | 0.1 |
| MC-Raba | 105.9 | 0.0 | 108.7 | 10.9 | 102.3 | 7.4 | 100.0 | 6.2 | 0.0 | 0.0 |
| MC-LA | 76.0 | 2.1 | 72.6 | 7.1 | 57.9 | 5.6 | 93.6 | 2.8 | 0.8 | 0.0 |
| MC-FA | 67.9 | 3.3 | 81.6 | 8.5 | 62.2 | 3.7 | 93.1 | 4.7 | 1.0 | 0.1 |
| MC-WA | 75.0 | 1.4 | 87.3 | 4.6 | 70.2 | 5.6 | 95.8 | 1.8 | 3.1 | 1.0 |
| MC-LAba | 69.2 | 5.8 | 86.5 | 7.1 | 68.1 | 4.0 | n.d. | n.d. | n.d. | n.d. |
| MC-FAba | 68.4 | 4.6 | 86.9 | 8.5 | 78.3 | 2.5 | n.d. | n.d. | n.d. | n.d. |
| MC-WAba | 71.9 | 3.0 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| MC-LF | n.d. | n.d. | n.d. | n.d. | 77.4 | 4.7 | n.d. | n.d. | n.d. | n.d. |
| D7-MC-LR | n.d. | n.d. | 97.8 | 6.6 | 92.6 | 5.9 | n.d. | n.d. | n.d. | n.d. |
| D5-MC-LF | n.d. | n.d. | 94.8 | 6.7 | 79.1 | 3.9 | n.d. | n.d. | n.d. | n.d. |
| Methanol | Acetonitrile | |||||||
|---|---|---|---|---|---|---|---|---|
| 1× | 2× | 1× | 2× | |||||
| Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| MC-RR | 43.2 | 2.1 | 52.3 | 5.0 | 44.6 | 2.6 | 47.1 | 2.8 |
| MC-YR | 35.1 | 2.2 | 47.4 | 7.1 | 43.8 | 4.2 | 47.2 | 3.0 |
| MC-LR | 43.8 | 2.3 | 49.4 | 5.2 | 45.9 | 2.8 | 46.7 | 2.6 |
| MC-FR | 44.3 | 2.3 | 56.3 | 6.0 | 50.8 | 2.9 | 52.1 | 3.5 |
| MC-WR | 41.0 | 3.0 | 54.0 | 5.6 | 45.6 | 3.3 | 48.0 | 3.2 |
| MC-RA | 50.9 | 2.2 | 62.0 | 7.5 | 54.8 | 2.5 | 55.7 | 2.7 |
| MC-Raba | 52.9 | 3.7 | 62.5 | 7.3 | 54.5 | 2.3 | 59.9 | 2.4 |
| MC-LA | 49.2 | 2.4 | 35.2 | 3.5 | 54.2 | 2.7 | 47.7 | 3.6 |
| MC-FA | 47.5 | 2.3 | 41.8 | 4.4 | 55.2 | 2.4 | 50.7 | 1.8 |
| MC-WA | 45.7 | 2.6 | 49.0 | 3.8 | 48.7 | 2.5 | 50.2 | 3.7 |
| MC-LAba | 49.7 | 2.8 | 47.0 | 6.0 | 57.7 | 3.0 | 56.6 | 3.3 |
| MC-FAba | 50.9 | 2.2 | 52.9 | 6.0 | 59.4 | 2.5 | 64.7 | 3.6 |
| MC-WAba | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| MC-LF | 59.2 | 5.3 | 61.1 | 5.8 | 69.0 | 6.4 | 64.3 | 6.2 |
| D7-MC-LR | 48.0 | 2.2 | 51.9 | 4.1 | 51.0 | 2.6 | 51.0 | 2.9 |
| D5-MC-LF | 61.0 | 2.5 | 67.4 | 6.0 | 70.0 | 3.1 | 71.6 | 3.3 |
| Mouse Recovery (% of Expected Result) | LOD (ng/mL) | LOQ (ng/mL) | Linear Range (ng/mL) | ||
|---|---|---|---|---|---|
| Congener | Mouse Serum | Mouse Liver | Mouse Serum | Mouse Serum | Mouse Serum |
| MC-RR | 74.2% | 78.7% | 0.5 | 2 | 2 - 500 |
| MC-YR | 88.5% | 82.9% | 0.5 | 2 | 2 - 500 |
| MC-LR | 113.1% | 81.2% | 0.1 | 2 | 2 - 500 |
| MC-FR | 124.3% | 86.3% | - | - | - |
| MC-WR | 108.6% | 94.5% | - | - | - |
| MC-RA | 111.2% | 120.9% | - | - | - |
| MC-RAba | 106.4% | 109.6% | - | - | - |
| MC-LA | 86.1% | 64.5% | - | - | - |
| MC-FA | 100.0% | 43.4% | - | - | - |
| MC-WA | 96.8% | 34.1% | - | - | - |
| MC-LAba | 94.3% | 42.2% | - | - | - |
| MC-FAba | 97.4% | 54.4% | - | - | - |
| MC-WAba | 89.8% | <LOQ | - | - | - |
| MC-LF | 83.5% | 90.6% | (0.5) | (2) | (2 - 500) |
| D7-MC-LR | 100.0% | 100.0% | - | - | - |
| D5-MC-LF | 100.0% | 100.0% | - | - | |
| Day 1 | Day 2 | Both Days | |||||
|---|---|---|---|---|---|---|---|
| Mean (ng/mL) | SD (ng/mL) | RSD (%) | Mean (ng/mL) | SD (ng/mL) | RSD (%) | RSD (%) | |
| MC-RR | 193.0 | 10.0 | 5.2 | 208.3 | 44.7 | 21.5 | 5.4 |
| MC-YR | 31.1 | 3.4 | 10.8 | 32.7 | 6.5 | 19.9 | 3.5 |
| MC-LR | 502.7 | 22.8 | 4.5 | 682.9 | 44.0 | 6.4 | 21.5 |
| MC-FR | 271.9 | 14.5 | 5.3 | 383.0 | 43.5 | 11.4 | 24.0 |
| MC-WR | 81.6 | 16.1 | 19.7 | 127.3 | 16.9 | 13.3 | 31.0 |
| MC-RA | 73.5 | 9.4 | 12.8 | 75.8 | 13.5 | 17.8 | 2.2 |
| MC-RAba | 21.1 | 1.0 | 4.5 | 22.6 | 2.7 | 12.1 | 5.0 |
| MC-LA | 112.6 | 17.9 | 15.9 | 102.9 | 8.6 | 8.3 | 6.4 |
| MC-FA | 145.5 | 2.4 | 1.7 | 119.6 | 6.0 | 5.0 | 13.8 |
| MC-WA | 46.3 | 3.1 | 6.7 | 38.0 | 5.5 | 14.6 | 14.0 |
| MC-LAba | 89.0 | 10.2 | 11.5 | 87.4 | 6.5 | 7.4 | 1.3 |
| MC-FAba | 49.7 | 8.1 | 16.2 | 39.1 | 3.1 | 8.0 | 16.9 |
| MC-WAba | <LOQ | n.d. | n.d. | < LOQ | n.d. | n.d. | n.d. |
| MC-LF | <LOQ | n.d. | n.d. | < LOQ | n.d. | n.d. | n.d. |
| D7-LR | 10.0 | 0.0 | 0.0 | 10.0 | 0.0 | 0.0 | 0.0 |
| D5-LF | 10.0 | 0.0 | 0.0 | 10.0 | 0.0 | 0.0 | 0.0 |
| Congener | Analysis Window (min) | Parent Mass (m/z) | Daughter Mass (m/z) | Dwell Time (s) | Cone Voltage (V) | Collision Energy (V) | External Standard | Internal Standard |
|---|---|---|---|---|---|---|---|---|
| MC-RR | 0.3–1.6 | 519.7 | 135.1 | 0.027 | 40 | 27 | MC-RR | D7-MC-LR |
| MC-YR | 0.3–1.6 | 1045.6 | 135.1 | 0.027 | 40 | 70 | MC-YR | D7-MC-LR |
| MC-LR | 0.3–1.6 | 995.6 | 135.1 | 0.027 | 40 | 65 | MC-LR | D7-MC-LR |
| MC-FR | 0.3–1.6 | 1029.6 | 135.1 | 0.027 | 40 | 65 | MC-LR | D7-MC-LR |
| MC-WR | 0.3–1.6 | 1086.6 | 135.1 | 0.027 | 40 | 65 | MC-LR | D7-MC-LR |
| MC-RA | 1.3–2.5 | 953.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D7-MC-LR |
| MC-Raba | 1.3–2.5 | 967.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D7-MC-LR |
| MC-LA | 1.3–2.5 | 910.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D7-MC-LF |
| MC-FA | 1.3–2.5 | 944.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D7-MC-LF |
| MC-WA | 1.3–2.5 | 983.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D5-MC-LF |
| MC-Laba | 1.7–3.5 | 924.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D5-MC-LF |
| MC-Faba | 1.7–3.5 | 958.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D5-MC-LF |
| MC-WAba | 1.7–3.5 | 997.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D5-MC-LF |
| MC-LF | 1.7–3.5 | 986.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | D5-MC-LF |
| D7-MC-LR | 1.0–1.8 | 1002.7 | 135.1 | 0.024 | 40 | 65 | MC-LR | - |
| D5-MC-LF | 2.3–3.5 | 991.6 | 135.1 | 0.024 | 40 | 65 | MC-LR | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altaner, S.; Puddick, J.; Fessard, V.; Feurstein, D.; Zemskov, I.; Wittmann, V.; Dietrich, D.R. Simultaneous Detection of 14 Microcystin Congeners from Tissue Samples Using UPLC- ESI-MS/MS and Two Different Deuterated Synthetic Microcystins as Internal Standards. Toxins 2019, 11, 388. https://doi.org/10.3390/toxins11070388
Altaner S, Puddick J, Fessard V, Feurstein D, Zemskov I, Wittmann V, Dietrich DR. Simultaneous Detection of 14 Microcystin Congeners from Tissue Samples Using UPLC- ESI-MS/MS and Two Different Deuterated Synthetic Microcystins as Internal Standards. Toxins. 2019; 11(7):388. https://doi.org/10.3390/toxins11070388
Chicago/Turabian StyleAltaner, Stefan, Jonathan Puddick, Valerie Fessard, Daniel Feurstein, Ivan Zemskov, Valentin Wittmann, and Daniel R. Dietrich. 2019. "Simultaneous Detection of 14 Microcystin Congeners from Tissue Samples Using UPLC- ESI-MS/MS and Two Different Deuterated Synthetic Microcystins as Internal Standards" Toxins 11, no. 7: 388. https://doi.org/10.3390/toxins11070388
APA StyleAltaner, S., Puddick, J., Fessard, V., Feurstein, D., Zemskov, I., Wittmann, V., & Dietrich, D. R. (2019). Simultaneous Detection of 14 Microcystin Congeners from Tissue Samples Using UPLC- ESI-MS/MS and Two Different Deuterated Synthetic Microcystins as Internal Standards. Toxins, 11(7), 388. https://doi.org/10.3390/toxins11070388