Mechanisms of Botulinum Toxin Type A Action on Pain


Abstract
:1. Introduction
2. Basic Pharmacology of BoNT/A: Mechanisms of Outstandingly High BoNT/A Potency and Long-Lasting Duration of Action
2.1. Structure of the BoNT/A Complex and Neurotoxin
2.2. Pharmacokinetics of Injected BoNT/A
2.3. Specificity of BoNT/A Effect: Acceptor-Mediated Entrance into the Neuronal Cytosol
2.4. Longevity of Light Chain-Mediated Enzymatic Activity
2.5. Inhibition of Neurotransmitter Release and the Effect on SNARE Supercomplex
2.6. Selectivity for Excitatory Synapses and Ca2+ Dynamics
2.7. Interaction with Ion Channels and Pain-Sensing Receptor Translocation
3. BoNT/A Effects on Peripheral Sensory Nerves
3.1. Prevention of Nociceptive Neurotransmitter Release in Peripheral Terminals
3.2. Anti-Inflammatory Effects of BoNT/A
3.3. Involvement of BoNT/A Systemic Effect in the Measurement of Nociceptive Responses
3.4. Regenerative Effects of BoNT/A in the Injured Nerve
3.5. Effects of BoNT/A on the Sensory Ganglia
4. Actions of BoNT/A in the Central Nervous System
4.1. Effects in TRPV1 Receptor-Expressing Central Afferent Terminals
4.2. Indirect Central Actions on the Endogenous Opioid and GABA Neurotransmission
4.3. Effects on Astroglia and Microglia (Neuroinflammation)
4.4. Effects on the Ascending Pain Processing Pathway
5. An Overview of Clinical Evidence of BoNT/A Analgesic Efficacy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arnon, S.S.; Schechter, R.; Inglesby, T.V.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Fine, A.D.; Hauer, J.; Layton, M.; et al. Botulinum toxin as a biological weapon: Medical and public health management. JAMA 2001, 285, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Burgen, A.S.V.; Dickens, F.; Zatman, L.J. The action of botulinum toxin on the neuro-muscular junction. J. Physiol. (Lond.) 1949, 109, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.B. Botulinum toxin injection into extraocular muscles as an alternative to strabismus surgery. J. Pediatr. Ophthalmol. Strabismus 1980, 17, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Safarpour, Y.; Jabbari, B. Botulinum Toxin Treatment of Movement Disorders. Curr. Treat. Options Neurol 2018, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. An update on new and unique uses of botulinum toxin in movement disorders. Toxicon 2018, 147, 84–88. [Google Scholar] [CrossRef]
- Tsui, J.K.; Eisen, A.; Stoessl, A.J.; Calne, S.; Calne, D.B. Double-blind study of botulinum toxin in spasmodic torticollis. Lancet 1986, 2, 245–247. [Google Scholar] [CrossRef]
- Cui, M.; Khanijou, S.; Rubino, J.; Aoki, K.R. Subcutaneous administration of botulinum toxin A reduces formalin-induced pain. Pain 2004, 107, 125–133. [Google Scholar] [CrossRef]
- Jackson, J.L.; Kuriyama, A.; Hayashino, Y. Botulinum toxin A for prophylactic treatment of migraine and tension headaches in adults: A meta-analysis. JAMA 2012, 307, 1736–1745. [Google Scholar] [CrossRef]
- Herd, C.P.; Tomlinson, C.L.; Rick, C.; Scotton, W.J.; Edwards, J.; Ives, N.; Clarke, C.E.; Sinclair, A. Botulinum toxins for the prevention of migraine in adults. Cochrane Database Syst. Rev. 2018, 2018, CD011616. [Google Scholar] [CrossRef]
- Dodick, D.W.; Turkel, C.C.; DeGryse, R.E.; Aurora, S.K.; Silberstein, S.D.; Lipton, R.B.; Diener, H.-C.; Brin, M.F. OnabotulinumtoxinA for Treatment of Chronic Migraine: Pooled Results From the Double-Blind, Randomized, Placebo-Controlled Phases of the PREEMPT Clinical Program. Headach J. Head Face Pain 2010, 50, 921–936. [Google Scholar] [CrossRef]
- Safarpour, Y.; Jabbari, B. Botulinum toxin treatment of pain syndromes—An evidence based review. Toxicon 2018, 147, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Tang-Liu, D.D.-S.; Aoki, K.R.; Dolly, J.O.; de Paiva, A.; Houchen, T.L.; Chasseaud, L.F.; Webber, C. Intramuscular injection of 125I-botulinum neurotoxin-complex versus 125I-botulinum-free neurotoxin: Time course of tissue distribution. Toxicon 2003, 42, 461–469. [Google Scholar] [CrossRef]
- Al-Saleem, F.H.; Nasser, Z.; Olson, R.M.; Cao, L.; Simpson, L.L. Identification of the Factors That Govern the Ability of Therapeutic Antibodies to Provide Postchallenge Protection Against Botulinum Toxin: A Model for Assessing Postchallenge Efficacy of Medical Countermeasures against Agents of Bioterrorism and Biological Warfare. J. Pharmacol. Exp. Ther. 2011, 338, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L. The life history of a botulinum toxin molecule. Toxicon 2013, 68, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleem, F.H.; Ancharski, D.M.; Joshi, S.G.; Elias, M.; Singh, A.; Nasser, Z.; Simpson, L.L. Analysis of the Mechanisms That Underlie Absorption of Botulinum Toxin by the Inhalation Route. Infect. Immun. 2012, 80, 4133–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.; Liu, S.; Acharya, K. Variations in the Botulinum Neurotoxin Binding Domain and the Potential for Novel Therapeutics. Toxins 2018, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S.; Bradshaw, M.; Tepp, W.H.; Pier, C.L.; Whitemarsh, R.C.M.; Chen, C.; Barbieri, J.T.; Johnson, E.A. The Light Chain Defines the Duration of Action of Botulinum Toxin Serotype A Subtypes. mBio 2018, 9, e00089-18. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Casals-Diaz, L.; Zurawski, T.; Meng, J.; Moriarty, O.; Nealon, J.; Edupuganti, O.P.; Dolly, O. A novel therapeutic with two SNAP-25 inactivating proteases shows long-lasting anti-hyperalgesic activity in a rat model of neuropathic pain. Neuropharmacology 2017, 118, 223–232. [Google Scholar] [CrossRef]
- Rossetto, O. The binding of botulinum neurotoxins to different peripheral neurons. Toxicon 2018, 147, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Blumenfeld, A.M.; Stark, R.J.; Freeman, M.C.; Orejudos, A.; Manack Adams, A. Long-term study of the efficacy and safety of OnabotulinumtoxinA for the prevention of chronic migraine: COMPEL study. J. Headache Pain 2018, 19, 13. [Google Scholar] [CrossRef]
- Matak, I.; Rossetto, O.; Lacković, Z. Botulinum toxin type A selectivity for certain types of pain is associated with capsaicin-sensitive neurons. Pain 2014, 155, 1516–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacković, Z.; Filipović, B.; Matak, I.; Helyes, Z. Activity of botulinum toxin type A in cranial dura: Implications for treatment of migraine and other headaches. Br. J. Pharmacol. 2016, 173, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Yao, L.; Ni, L.; Wang, L.; Hu, X. Antinociceptive effect of botulinum toxin A involves alterations in AMPA receptor expression and glutamate release in spinal dorsal horn neurons. Neuroscience 2017, 357, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Drinovac Vlah, V.; Bach-Rojecky, L.; Lacković, Z. Antinociceptive action of botulinum toxin type A in carrageenan-induced mirror pain. J. Neural Transm. (Vienna) 2016, 123, 1403–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.H.; Shim, J.H.; Yeom, J.H.; Shin, W.J.; Cho, S.Y.; Jeon, W.J. Ultrasound-guided greater occipital nerve block with botulinum toxin for patients with chronic headache in the occipital area: A randomized controlled trial. Korean J. Anesthesiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Frießem, C.H.; Eitner, L.B.; Kaisler, M.; Maier, C.; Vollert, J.; Westermann, A.; Zahn, P.K.; Avila González, C.A. Perineural injection of botulinum toxin-A in painful peripheral nerve injury—A case series: Pain relief, safety, sensory profile and sample size recommendation. Curr. Med. Res. Opin. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Drinovac Vlah, V.; Bach-Rojecky, L.; Matak, I.; Lacković, Z. Involvement of μ-opioid receptors in antinociceptive action of botulinum toxin type A. Neuropharmacology 2013, 70, 331–337. [Google Scholar] [CrossRef]
- Sandrini, G.; Perrotta, A.; Tassorelli, C.; Torelli, P.; Brighina, F.; Sances, G.; Nappi, G. Botulinum toxin type-A in the prophylactic treatment of medication-overuse headache: A multicenter, double-blind, randomized, placebo-controlled, parallel group study. J. Headache Pain 2011, 12, 427–433. [Google Scholar] [CrossRef]
- Butera, C.; Colombo, B.; Bianchi, F.; Cursi, M.; Messina, R.; Amadio, S.; Guerriero, R.; Comi, G.; Del Carro, U. Refractory chronic migraine: Is drug withdrawal necessary before starting a therapy with onabotulinum toxin type A? Neurol. Sci. 2016, 37, 1701–1706. [Google Scholar] [CrossRef]
- Ghosal, K.J.; Patel, K.; Singh, B.R.; Hale, M.L. Role of critical elements in botulinum neurotoxin complex in toxin routing across intestinal and bronchial barriers. PLoS ONE 2018, 13, e0199524. [Google Scholar] [CrossRef]
- Eisele, K.-H.; Fink, K.; Vey, M.; Taylor, H.V. Studies on the dissociation of botulinum neurotoxin type A complexes. Toxicon 2011, 57, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D.; Pan, L.; Bigalke, H. Comparing incobotulinumtoxinA (Xeomin®) and onabotulinumtoxinA (Botox®): Identical potency labelling in the hemidiaphragm assay. J. Neural Transm. 2018, 125, 1351–1354. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed]
- Ranoux, D.; Attal, N.; Morain, F.; Bouhassira, D. Botulinum toxin type a induces direct analgesic effects in chronic neuropathic pain. Ann. Neurol. 2008, 64, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.-Y.; Park, J.-H.; Youn, C.S.; Lee, J.H. Intradermal Injection of Botulinum Toxin: A Safer Treatment Modality for Forehead Wrinkles. Ann. Dermatol. 2018, 30, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Muraro, L.; Tosatto, S.; Motterlini, L.; Rossetto, O.; Montecucco, C. The N-terminal half of the receptor domain of botulinum neurotoxin A binds to microdomains of the plasma membrane. Biochem. Biophys. Res. Commun. 2009, 380, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Martin, S.; Papadopulos, A.; Harper, C.B.; Mavlyutov, T.A.; Niranjan, D.; Glass, N.R.; Cooper-White, J.J.; Sibarita, J.-B.; Choquet, D.; et al. Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. J. Neurosci. 2015, 35, 6179–6194. [Google Scholar] [CrossRef]
- Restani, L.; Giribaldi, F.; Manich, M.; Bercsenyi, K.; Menendez, G.; Rossetto, O.; Caleo, M.; Schiavo, G. Botulinum neurotoxins A and E undergo retrograde axonal transport in primary motor neurons. PLoS Pathog. 2012, 8, e1003087. [Google Scholar] [CrossRef] [PubMed]
- Jacky, B.P.S.; Garay, P.E.; Dupuy, J.; Nelson, J.B.; Cai, B.; Molina, Y.; Wang, J.; Steward, L.E.; Broide, R.S.; Francis, J.; et al. Identification of fibroblast growth factor receptor 3 (FGFR3) as a protein receptor for botulinum neurotoxin serotype A (BoNT/A). PLoS Pathog. 2013, 9, e1003369. [Google Scholar] [CrossRef]
- Li, X.; Coffield, J.A. Structural and Functional Interactions between Transient Receptor Potential Vanilloid Subfamily 1 and Botulinum Neurotoxin Serotype A. PLoS ONE 2016, 11, e0143024. [Google Scholar] [CrossRef]
- Vagin, O.; Beenhouwer, D.O. Septins: Regulators of Protein Stability. Front. Cell Dev. Biol. 2016, 4, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, C.B.; Oyler, G.A. Persistence of Botulinum neurotoxin inactivation of nerve function. Curr. Top. Microbiol. Immunol. 2013, 364, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Whitemarsh, R.C.M.; Tepp, W.H.; Johnson, E.A.; Pellett, S. Persistence of botulinum neurotoxin a subtypes 1-5 in primary rat spinal cord cells. PLoS ONE 2014, 9, e90252. [Google Scholar] [CrossRef] [PubMed]
- Kutschenko, A.; Reinert, M.-C.; Krez, N.; Liebetanz, D.; Rummel, A. BoNT/AB hybrid maintains similar duration of paresis as BoNT/A wild-type in murine running wheel assay. Neurotoxicology 2017, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Relja, M.; Lacković, Z. Botulinum toxin type A in experimental neuropathic pain. J. Neural Transm. (Vienna) 2005, 112, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Dai, J.; Zhang, D. Botulinum toxin decreases hyperalgesia and inhibits P2X3 receptor over-expression in sensory neurons induced by ventral root transection in rats. Pain Med. 2011, 12, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Raciborska, D.A.; Charlton, M.P. Retention of cleaved synaptosome-associated protein of 25 kDa (SNAP-25) in neuromuscular junctions: A new hypothesis to explain persistence of botulinum A poisoning. Can. J. Physiol. Pharmacol. 1999, 77, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Bartels, F.; Bergel, H.; Bigalke, H.; Frevert, J.; Halpern, J.; Middlebrook, J. Specific antibodies against the Zn(2+)-binding domain of clostridial neurotoxins restore exocytosis in chromaffin cells treated with tetanus or botulinum A neurotoxin. J. Biol. Chem. 1994, 269, 8122–8127. [Google Scholar]
- Keller, J.E.; Neale, E.A. The role of the synaptic protein snap-25 in the potency of botulinum neurotoxin type A. J. Biol. Chem. 2001, 276, 13476–13482. [Google Scholar] [CrossRef]
- Bajohrs, M.; Rickman, C.; Binz, T.; Davletov, B. A molecular basis underlying differences in the toxicity of botulinum serotypes A and E. EMBO Rep. 2004, 5, 1090–1095. [Google Scholar] [CrossRef]
- Huang, X.; Wheeler, M.B.; Kang, Y.H.; Sheu, L.; Lukacs, G.L.; Trimble, W.S.; Gaisano, H.Y. Truncated SNAP-25 (1-197), like botulinum neurotoxin A, can inhibit insulin secretion from HIT-T15 insulinoma cells. Mol. Endocrinol. 1998, 12, 1060–1070. [Google Scholar] [CrossRef] [PubMed]
- Jurasinski, C.V.; Lieth, E.; Dang Do, A.N.; Schengrund, C.L. Correlation of cleavage of SNAP-25 with muscle function in a rat model of Botulinum neurotoxin type A induced paralysis. Toxicon 2001, 39, 1309–1315. [Google Scholar] [CrossRef]
- Lawrence, G.W.; Ovsepian, S.V.; Wang, J.; Aoki, K.R.; Dolly, J.O. Extravesicular intraneuronal migration of internalized botulinum neurotoxins without detectable inhibition of distal neurotransmission. Biochem. J. 2012, 441, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Megighian, A.; Scorzeto, M.; Zanini, D.; Pantano, S.; Rigoni, M.; Benna, C.; Rossetto, O.; Montecucco, C.; Zordan, M. Arg206 of SNAP-25 is essential for neuroexocytosis at the Drosophila melanogaster neuromuscular junction. J. Cell. Sci. 2010, 123, 3276–3283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakov, R.; Habermann, E.; Hertting, G.; Wurster, S.; Allgaier, C. Effects of botulinum A toxin on presynaptic modulation of evoked transmitter release. Eur. J. Pharmacol. 1989, 164, 45–53. [Google Scholar] [CrossRef]
- McMahon, H.T.; Foran, P.; Dolly, J.O.; Verhage, M.; Wiegant, V.M.; Nicholls, D.G. Tetanus toxin and botulinum toxins type A and B inhibit glutamate, gamma-aminobutyric acid, aspartate, and met-enkephalin release from synaptosomes. Clues to the locus of action. J. Biol. Chem. 1992, 267, 21338–21343. [Google Scholar] [PubMed]
- Südhof, T.C. A molecular machine for neurotransmitter release: Synaptotagmin and beyond. Nat. Med. 2013, 19, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Gerona, R.R.; Larsen, E.C.; Kowalchyk, J.A.; Martin, T.F. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J. Biol. Chem. 2000, 275, 6328–6336. [Google Scholar] [CrossRef]
- Meng, J.; Wang, J.; Lawrence, G.; Dolly, J.O. Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J. Cell. Sci. 2007, 120, 2864–2874. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Ovsepian, S.V.; Wang, J.; Pickering, M.; Sasse, A.; Aoki, K.R.; Lawrence, G.W.; Dolly, J.O. Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J. Neurosci. 2009, 29, 4981–4992. [Google Scholar] [CrossRef]
- Durham, P.L.; Cady, R.; Cady, R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache 2004, 44, 35–42; discussion 42–43. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Grumelli, C.; Raiteri, L.; Coco, S.; Paluzzi, S.; Caccin, P.; Rossetto, O.; Bonanno, G.; Montecucco, C.; Matteoli, M. Traffic of botulinum toxins A and E in excitatory and inhibitory neurons. Traffic 2007, 8, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Condliffe, S.; Bozzi, Y.; Chikhladze, M.; Grumelli, C.; Proux-Gillardeaux, V.; Takahashi, M.; Franceschetti, S.; Verderio, C.; Matteoli, M. Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc. Natl. Acad. Sci. USA 2008, 105, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumelli, C.; Corradini, I.; Matteoli, M.; Verderio, C. Intrinsic calcium dynamics control botulinum toxin A susceptibility in distinct neuronal populations. Cell Calcium 2010, 47, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Morenilla-Palao, C.; Planells-Cases, R.; García-Sanz, N.; Ferrer-Montiel, A. Regulated Exocytosis Contributes to Protein Kinase C Potentiation of Vanilloid Receptor Activity. J. Biol. Chem. 2004, 279, 25665–25672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Shibata, M.; Toriumi, H.; Iwashita, T.; Funakubo, M.; Sato, H.; Kuroi, T.; Ebine, T.; Koizumi, K.; Suzuki, N. Reduction of TRPV1 expression in the trigeminal system by botulinum neurotoxin type-A. Neurobiol. Dis. 2012, 48, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.-C.; Wakita, M.; Xie, D.-J.; Yamaga, T.; Iwata, S.; Torii, Y.; Harakawa, T.; Ginnaga, A.; Kozaki, S.; Akaike, N. Inhibition of membrane Na+ channels by A type botulinum toxin at femtomolar concentrations in central and peripheral neurons. J. Pharmacol. Sci. 2012, 118, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Zhang, X.; Levy, D.; Aoki, K.R.; Brin, M.F. Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: Therapeutic implications for migraine and other pains. Cephalalgia 2014, 34, 853–869. [Google Scholar] [CrossRef] [PubMed]
- Paterson, K.; Lolignier, S.; Wood, J.N.; McMahon, S.B.; Bennett, D.L.H. Botulinum toxin-A treatment reduces human mechanical pain sensitivity and mechanotransduction. Ann. Neurol. 2014, 75, 591–596. [Google Scholar] [CrossRef]
- Purkiss, J.; Welch, M.; Doward, S.; Foster, K. Capsaicin-stimulated release of substance P from cultured dorsal root ganglion neurons: Involvement of two distinct mechanisms. Biochem. Pharmacol. 2000, 59, 1403–1406. [Google Scholar] [CrossRef]
- Rapp, D.E.; Turk, K.W.; Bales, G.T.; Cook, S.P. Botulinum toxin type a inhibits calcitonin gene-related peptide release from isolated rat bladder. J. Urol. 2006, 175, 1138–1142. [Google Scholar] [CrossRef]
- Lucioni, A.; Bales, G.T.; Lotan, T.L.; McGehee, D.S.; Cook, S.P.; Rapp, D.E. Botulinum toxin type A inhibits sensory neuropeptide release in rat bladder models of acute injury and chronic inflammation. BJU Int. 2008, 101, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Lora, V.R.M.M.; Clemente-Napimoga, J.T.; Abdalla, H.B.; Macedo, C.G.; de la Torre Canales, G.; Barbosa, C.M.R. Botulinum toxin type A reduces inflammatory hypernociception induced by arthritis in the temporomadibular joint of rats. Toxicon 2017, 129, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt da Silva, L.; Karshenas, A.; Bach, F.W.; Rasmussen, S.; Arendt-Nielsen, L.; Gazerani, P. Blockade of glutamate release by botulinum neurotoxin type A in humans: A dermal microdialysis study. Pain Res. Manag. 2014, 19, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Dominis, M.; Lacković, Z. Lack of anti-inflammatory effect of botulinum toxin type A in experimental models of inflammation. Fundam. Clin. Pharmacol. 2008, 22, 503–509. [Google Scholar] [CrossRef]
- Chuang, Y.-C.; Yoshimura, N.; Huang, C.-C.; Wu, M.; Chiang, P.-H.; Chancellor, M.B. Intraprostatic botulinum toxin a injection inhibits cyclooxygenase-2 expression and suppresses prostatic pain on capsaicin induced prostatitis model in rat. J. Urol. 2008, 180, 742–748. [Google Scholar] [CrossRef]
- Chuang, Y.-C.; Yoshimura, N.; Huang, C.-C.; Wu, M.; Chiang, P.-H.; Chancellor, M.B. Intravesical botulinum toxin A administration inhibits COX-2 and EP4 expression and suppresses bladder hyperactivity in cyclophosphamide-induced cystitis in rats. Eur. Urol. 2009, 56, 159–166. [Google Scholar] [CrossRef]
- Yoo, K.Y.; Lee, H.S.; Cho, Y.K.; Lim, Y.S.; Kim, Y.S.; Koo, J.H.; Yoon, S.J.; Lee, J.H.; Jang, K.H.; Song, S.H. Anti-inflammatory effects of botulinum toxin type a in a complete Freund’s adjuvant-induced arthritic knee joint of hind leg on rat model. Neurotox. Res. 2014, 26, 32–39. [Google Scholar] [CrossRef]
- Wang, L.; Wang, K.; Chu, X.; Li, T.; Shen, N.; Fan, C.; Niu, Z.; Zhang, X.; Hu, L. Intra-articular injection of Botulinum toxin A reduces neurogenic inflammation in CFA-induced arthritic rat model. Toxicon 2017, 126, 70–78. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, Y.; Lee, J.; Park, C.; Moon, D.E. The effects of botulinum toxin A on mechanical and cold allodynia in a rat model of neuropathic pain. Can. J. Anaesth. 2006, 53, 470–477. [Google Scholar] [CrossRef] [Green Version]
- Luvisetto, S.; Marinelli, S.; Cobianchi, S.; Pavone, F. Anti-allodynic efficacy of botulinum neurotoxin A in a model of neuropathic pain. Neuroscience 2007, 145, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Favre-Guilmard, C.; Auguet, M.; Chabrier, P.-E. Different antinociceptive effects of botulinum toxin type A in inflammatory and peripheral polyneuropathic rat models. Eur. J. Pharmacol. 2009, 617, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Salković-Petrisić, M.; Lacković, Z. Botulinum toxin type A reduces pain supersensitivity in experimental diabetic neuropathy: Bilateral effect after unilateral injection. Eur. J. Pharmacol. 2010, 633, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Marinelli, S.; Luvisetto, S.; Cobianchi, S.; Makuch, W.; Obara, I.; Mezzaroma, E.; Caruso, M.; Straface, E.; Przewlocka, B.; Pavone, F. Botulinum neurotoxin type A counteracts neuropathic pain and facilitates functional recovery after peripheral nerve injury in animal models. Neuroscience 2010, 171, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Mika, J.; Rojewska, E.; Makuch, W.; Korostynski, M.; Luvisetto, S.; Marinelli, S.; Pavone, F.; Przewlocka, B. The effect of botulinum neurotoxin A on sciatic nerve injury-induced neuroimmunological changes in rat dorsal root ganglia and spinal cord. Neuroscience 2011, 175, 358–366. [Google Scholar] [CrossRef]
- Filipović, B.; Matak, I.; Bach-Rojecky, L.; Lacković, Z. Central Action of Peripherally Applied Botulinum Toxin Type A on Pain and Dural Protein Extravasation in Rat Model of Trigeminal Neuropathy. PLoS ONE 2012, 7, e29803. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Zhuang, Y.; Qu, W.; Muir, J.; Liang, H.; Zhang, D. Botulinum toxin type A reduces hyperalgesia and TRPV1 expression in rats with neuropathic pain. Pain Med. 2013, 14, 276–286. [Google Scholar] [CrossRef]
- Vacca, V.; Marinelli, S.; Luvisetto, S.; Pavone, F. Botulinum toxin A increases analgesic effects of morphine, counters development of morphine tolerance and modulates glia activation and μ opioid receptor expression in neuropathic mice. Brain Behav. Immun. 2013, 32, 40–50. [Google Scholar] [CrossRef]
- Drinovac Vlah, V.; Bach-Rojecky, L.; Lacković, Z. Association of antinociceptive action of botulinum toxin type A with GABA-A receptor. J. Neural Transm. (Vienna) 2014, 121, 665–669. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Xie, N.; Lian, Y.; Xu, H.; Chen, C.; Zheng, Y.; Chen, Y.; Zhang, H. Central antinociceptive activity of peripherally applied botulinum toxin type A in lab rat model of trigeminal neuralgia. Springerplus 2016, 5, 431. [Google Scholar] [CrossRef]
- Piovesan, E.J.; Oshinsky, M.; Silberstein, S.; Kowacs, P.A.; Novak, E.M.; Werneck, L.C. Botulinum neurotoxin type-A when utilized in animals with trigeminal sensitization induced a antinociceptive effect. Arq. Neuropsiquiatr. 2016, 74, 462–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.Y.; Kim, M.J.; Ju, J.S.; Park, S.K.; Lee, C.G.; Kim, S.T.; Bae, Y.C.; Ahn, D.K. Antinociceptive Effects of Botulinum Toxin Type A on Trigeminal Neuropathic Pain. J. Dent. Res. 2016, 95, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Zychowska, M.; Rojewska, E.; Makuch, W.; Luvisetto, S.; Pavone, F.; Marinelli, S.; Przewlocka, B.; Mika, J. Participation of pro- and anti-nociceptive interleukins in botulinum toxin A-induced analgesia in a rat model of neuropathic pain. Eur. J. Pharmacol. 2016, 791, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Favre-Guilmard, C.; Chabrier, P.-E.; Kalinichev, M. Bilateral analgesic effects of abobotulinumtoxinA (Dysport®) following unilateral administration in the rat. Eur. J. Pain 2017, 21, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Tékus, V.; Bölcskei, K.; Lacković, Z.; Helyes, Z. Involvement of substance P in the antinociceptive effect of botulinum toxin type A: Evidence from knockout mice. Neuroscience 2017, 358, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, S.; Vacca, V.; Ricordy, R.; Uggenti, C.; Tata, A.M.; Luvisetto, S.; Pavone, F. The analgesic effect on neuropathic pain of retrogradely transported botulinum neurotoxin A involves Schwann cells and astrocytes. PLoS ONE 2012, 7, e47977. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, A.; Marinelli, S.; De Angelis, F.; Vacca, V.; Luvisetto, S.; Pavone, F. Botulinum Toxin B Affects Neuropathic Pain but Not Functional Recovery after Peripheral Nerve Injury in a Mouse Model. Toxins (Basel) 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Matsuka, Y.; Spigelman, I.; Ishihara, Y.; Yamamoto, Y.; Sonoyama, W.; Kamioka, H.; Yamashiro, T.; Kuboki, T.; Oguma, K. Botulinum toxin type a (150 kDa) decreases exaggerated neurotransmitter release from trigeminal ganglion neurons and relieves neuropathy behaviors induced by infraorbital nerve constriction. Neuroscience 2009, 159, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Lam, C.; Yaksh, T.L. Botulinum toxin in migraine: Role of transport in trigemino-somatic and trigemino-vascular afferents. Neurobiol. Dis. 2015, 79, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittencourt da Silva, L.; Poulsen, J.N.; Arendt-Nielsen, L.; Gazerani, P. Botulinum neurotoxin type A modulates vesicular release of glutamate from satellite glial cells. J. Cell Mol. Med. 2015, 19, 1900–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matak, I.; Bach-Rojecky, L.; Filipović, B.; Lacković, Z. Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience 2011, 186, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, R.; Bement, M.K.H.; Skyba, D.; Sluka, K.A.; Kehl, L.J. Models of muscle pain: Carrageenan model and acidic saline model. Curr. Protoc. Pharmacol. 2004. Chapter 5 Unit 5.35. [Google Scholar] [CrossRef]
- Da Silva, L.F.; Desantana, J.M.; Sluka, K.A. Activation of NMDA receptors in the brainstem, rostral ventromedial medulla, and nucleus reticularis gigantocellularis mediates mechanical hyperalgesia produced by repeated intramuscular injections of acidic saline in rats. J. Pain 2010, 11, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Lacković, Z. Central origin of the antinociceptive action of botulinum toxin type A. Pharmacol. Biochem. Behav. 2009, 94, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matak, I.; Riederer, P.; Lacković, Z. Botulinum toxin’s axonal transport from periphery to the spinal cord. Neurochem. Int. 2012, 61, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Anand, U.; Otto, W.R.; Sinisi, M.; Fox, M.; Birch, R.; Foster, K.A.; Mukerji, G.; Akbar, A.; Agarwal, S.K.; et al. Increased levels of SV2A botulinum neurotoxin receptor in clinical sensory disorders and functional effects of botulinum toxins A and E in cultured human sensory neurons. J. Pain Res. 2011, 4, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attal, N.; de Andrade, D.C.; Adam, F.; Ranoux, D.; Teixeira, M.J.; Galhardoni, R.; Raicher, I.; Üçeyler, N.; Sommer, C.; Bouhassira, D. Safety and efficacy of repeated injections of botulinum toxin A in peripheral neuropathic pain (BOTNEP): A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2016, 15, 555–565. [Google Scholar] [CrossRef]
- Drinovac Vlah, V.; Filipović, B.; Bach-Rojecky, L.; Lacković, Z. Role of central versus peripheral opioid system in antinociceptive and anti-inflammatory effect of botulinum toxin type A in trigeminal region. Eur. J. Pain 2018, 22, 583–591. [Google Scholar] [CrossRef]
- Vacca, V.; Marinelli, S.; Eleuteri, C.; Luvisetto, S.; Pavone, F. Botulinum neurotoxin A enhances the analgesic effects on inflammatory pain and antagonizes tolerance induced by morphine in mice. Brain Behav. Immun. 2012, 26, 489–499. [Google Scholar] [CrossRef]
- Chen, S.-R.; Prunean, A.; Pan, H.-M.; Welker, K.L.; Pan, H.-L. Resistance to morphine analgesic tolerance in rats with deleted transient receptor potential vanilloid type 1-expressing sensory neurons. Neuroscience 2007, 145, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Geis, C.; Sommer, C. Activation of TRPV1 contributes to morphine tolerance: Involvement of the mitogen-activated protein kinase signaling pathway. J. Neurosci. 2008, 28, 5836–5845. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-L.; Chen, S.-R.; Chen, H.; Pan, H.-L. Chronic opioid potentiates presynaptic but impairs postsynaptic N-methyl-D-aspartic acid receptor activity in spinal cords: Implications for opioid hyperalgesia and tolerance. J. Biol. Chem. 2012, 287, 25073–25085. [Google Scholar] [CrossRef] [PubMed]
- Hull, L.C.; Gabra, B.H.; Bailey, C.P.; Henderson, G.; Dewey, W.L. Reversal of morphine analgesic tolerance by ethanol in the mouse. J. Pharmacol. Exp. Ther. 2013, 345, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, A.; Popiolek-Barczyk, K.; Pavone, F.; Mika, J. Comparison of the Expression Changes after Botulinum Toxin Type A and Minocycline Administration in Lipopolysaccharide-Stimulated Rat Microglial and Astroglial Cultures. Front. Cell Infect. Microbiol. 2017, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Kim, J.-H.; Lee, K.-J.; Choi, M.-M.; Kim, Y.H.; Rhie, G.-E.; Yoo, C.-K.; Cha, K.; Shin, N.-R. Botulinum neurotoxin type A induces TLR2-mediated inflammatory responses in macrophages. PLoS ONE 2015, 10, e0120840. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Antonucci, F.; Gianfranceschi, L.; Rossi, C.; Rossetto, O.; Caleo, M. Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A). J. Neurosci. 2011, 31, 15650–15659. [Google Scholar] [CrossRef]
- Restani, L.; Novelli, E.; Bottari, D.; Leone, P.; Barone, I.; Galli-Resta, L.; Strettoi, E.; Caleo, M. Botulinum neurotoxin A impairs neurotransmission following retrograde transynaptic transport. Traffic 2012, 13, 1083–1089. [Google Scholar] [CrossRef]
- Koizumi, H.; Goto, S.; Okita, S.; Morigaki, R.; Akaike, N.; Torii, Y.; Harakawa, T.; Ginnaga, A.; Kaji, R. Spinal Central Effects of Peripherally Applied Botulinum Neurotoxin A in Comparison between Its Subtypes A1 and A2. Front. Neurol. 2014, 5, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibragić, S.; Matak, I.; Dračić, A.; Smajlović, A.; Muminović, M.; Proft, F.; Sofić, E.; Lacković, Z.; Riederer, P. Effects of botulinum toxin type A facial injection on monoamines and their metabolites in sensory, limbic and motor brain regions in rats. Neurosci. Lett. 2016, 617, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Khavari, R.; Elias, S.N.; Pande, R.; Wu, K.M.; Boone, T.B.; Karmonik, C. Higher Neural Correlates in Patients with Multiple Sclerosis and Neurogenic Overactive Bladder Following Treatment with Intradetrusor Injection of OnabotulinumtoxinA. J. Urol. 2019, 201, 135–140. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Loder, E.; Rizzoli, P. Pharmacologic Prevention of Migraine: A Narrative Review of the State of the Art in 2018. Headache J. Head Face Pain 2018, 58, 218–229. [Google Scholar] [CrossRef]
- Botz, B.; Bölcskei, K.; Helyes, Z. Challenges to develop novel anti-inflammatory and analgesic drugs. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1427. [Google Scholar] [CrossRef]
- Blersch, W.; Schulte-Mattler, W.J.; Przywara, S.; May, A.; Bigalke, H.; Wohlfarth, K. Botulinum toxin A and the cutaneous nociception in humans: A prospective, double-blind, placebo-controlled, randomized study. J. Neurol. Sci. 2002, 205, 59–63. [Google Scholar] [CrossRef]
- Voller, B.; Sycha, T.; Gustorff, B.; Schmetterer, L.; Lehr, S.; Eichler, H.G.; Auff, E.; Schnider, P. A randomized, double-blind, placebo controlled study on analgesic effects of botulinum toxin A. Neurology 2003, 61, 940–944. [Google Scholar] [CrossRef]
- Schulte-Mattler, W.J.; Opatz, O.; Blersch, W.; May, A.; Bigalke, H.; Wohlfahrt, K. Botulinum toxin A does not alter capsaicin-induced pain perception in human skin. J. Neurol. Sci. 2007, 260, 38–42. [Google Scholar] [CrossRef]
- Gazerani, P.; Staahl, C.; Drewes, A.M.; Arendt-Nielsen, L. The effects of Botulinum Toxin type A on capsaicin-evoked pain, flare, and secondary hyperalgesia in an experimental human model of trigeminal sensitization. Pain 2006, 122, 315–325. [Google Scholar] [CrossRef]
- Gazerani, P.; Pedersen, N.S.; Staahl, C.; Drewes, A.M.; Arendt-Nielsen, L. Subcutaneous Botulinum toxin type A reduces capsaicin-induced trigeminal pain and vasomotor reactions in human skin. Pain 2009, 141, 60–69. [Google Scholar] [CrossRef]
- Hepp, Z.; Dodick, D.W.; Varon, S.F.; Gillard, P.; Hansen, R.N.; Devine, E.B. Adherence to oral migraine-preventive medications among patients with chronic migraine. Cephalalgia 2015, 35, 478–488. [Google Scholar] [CrossRef]
- Hepp, Z.; Dodick, D.W.; Varon, S.F.; Chia, J.; Matthew, N.; Gillard, P.; Hansen, R.N.; Devine, E.B. Persistence and switching patterns of oral migraine prophylactic medications among patients with chronic migraine: A retrospective claims analysis. Cephalalgia 2017, 37, 470–485. [Google Scholar] [CrossRef]
- Pijpers, J.A.; Kies, D.A.; Louter, M.A.; van Zwet, E.W.; Ferrari, M.D.; Terwindt, G.M. Acute withdrawal and botulinum toxin A in chronic migraine with medication overuse: A double-blind randomized controlled trial. Brain 2019, 142, 1203–1214. [Google Scholar] [CrossRef]
- Jakubowski, M.; McAllister, P.J.; Bajwa, Z.H.; Ward, T.N.; Smith, P.; Burstein, R. Exploding vs. imploding headache in migraine prophylaxis with Botulinum Toxin A. Pain 2006, 125, 286–295. [Google Scholar] [CrossRef]
- Burstein, R.; Dodick, D.; Silberstein, S. Migraine prophylaxis with botulinum toxin A is associated with perception of headache. Toxicon 2009, 54, 624–627. [Google Scholar] [CrossRef] [Green Version]
- Cernuda-Morollõn, E.; Martínez-Camblor, P.; Ramõn, C.; Larrosa, D.; Serrano-Pertierra, E.; Pascual, J. CGRP and VIP levels as predictors of efficacy of onabotulinumtoxin type A in chronic migraine. Headache 2014, 54, 987–995. [Google Scholar] [CrossRef]
- Cernuda-Morollón, E.; Ramón, C.; Martínez-Camblor, P.; Serrano-Pertierra, E.; Larrosa, D.; Pascual, J. OnabotulinumtoxinA decreases interictal CGRP plasma levels in patients with chronic migraine. PAIN 2015, 156, 820–824. [Google Scholar] [CrossRef]
- Young, W.B.; Ivan Lopez, J.; Rothrock, J.F.; Orejudos, A.; Manack Adams, A.; Lipton, R.B.; Blumenfeld, A.M. Effects of onabotulinumtoxinA treatment in patients with and without allodynia: Results of the COMPEL study. J. Headache Pain 2019, 20, 10. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, C.; Choi, H.; Chung, C.-S. Factors associated with favorable outcome in botulinum toxin A treatment for chronic migraine: A clinic-based prospective study. J. Neurol. Sci. 2016, 363, 51–54. [Google Scholar] [CrossRef]
- Shackleton, T.; Ram, S.; Black, M.; Ryder, J.; Clark, G.T.; Enciso, R. The efficacy of botulinum toxin for the treatment of trigeminal and postherpetic neuralgia: A systematic review with meta-analyses. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2016, 122, 61–71. [Google Scholar] [CrossRef]
- Meng, F.; Peng, K.; Yang, J.-P.; Ji, F.-H.; Xia, F.; Meng, X.-W. Botulinum toxin-A for the treatment of neuralgia: A systematic review and meta-analysis. J. Pain Res. 2018, 11, 2343–2351. [Google Scholar] [CrossRef]
- A Study Evaluating Botulinum Toxin Type A in Subjects with Postherpetic Neuralgia—Study Synopsis. Available online: https://www.allerganclinicaltrials.com/en/trial-details/?id=191622-066 (accessed on 1 July 2019).
- Xiao, L.; Mackey, S.; Hui, H.; Xong, D.; Zhang, Q.; Zhang, D. Subcutaneous Injection of Botulinum Toxin A Is Beneficial in Postherpetic Neuralgia. Pain Med. 2010, 11, 1827–1833. [Google Scholar] [CrossRef] [Green Version]
- Apalla, Z.; Sotiriou, E.; Lallas, A.; Lazaridou, E.; Ioannides, D. Botulinum toxin A in postherpetic neuralgia: A parallel, randomized, double-blind, single-dose, placebo-controlled trial. Clin. J. Pain 2013, 29, 857–864. [Google Scholar] [CrossRef]
- Wu, C.-J.; Lian, Y.-J.; Zheng, Y.-K.; Zhang, H.-F.; Chen, Y.; Xie, N.-C.; Wang, L.-J. Botulinum toxin type A for the treatment of trigeminal neuralgia: Results from a randomized, double-blind, placebo-controlled trial. Cephalalgia 2012, 32, 443–450. [Google Scholar] [CrossRef]
- Shehata, H.S.; El-Tamawy, M.S.; Shalaby, N.M.; Ramzy, G. Botulinum toxin-type A: Could it be an effective treatment option in intractable trigeminal neuralgia? J. Headache Pain 2013, 14, 92. [Google Scholar] [CrossRef]
- Zúñiga, C.; Piedimonte, F.; Díaz, S.; Micheli, F. Acute Treatment of Trigeminal Neuralgia With Onabotulinum Toxin A. Clin. Neuropharmacol. 2013, 36, 146–150. [Google Scholar] [CrossRef]
- Zhang, H.; Lian, Y.; Ma, Y.; Chen, Y.; He, C.; Xie, N.; Wu, C. Two doses of botulinum toxin type A for the treatment of trigeminal neuralgia: Observation of therapeutic effect from a randomized, double-blind, placebo-controlled trial. J. Headache Pain 2014, 15, 65. [Google Scholar] [CrossRef]
- Yuan, R.Y.; Sheu, J.J.; Yu, J.M.; Chen, W.T.; Tseng, I.J.; Chang, H.H.; Hu, C.J. Botulinum toxin for diabetic neuropathic pain: A randomized double-blind crossover trial. Neurology 2009, 72, 1473–1478. [Google Scholar] [CrossRef]
- Zhang, T.; Adatia, A.; Zarin, W.; Moitri, M.; Vijenthira, A.; Chu, R.; Thabane, L.; Kean, W. The efficacy of botulinum toxin type A in managing chronic musculoskeletal pain: A systematic review and meta analysis. Inflammopharmacology 2011, 19, 21–34. [Google Scholar] [CrossRef]
- Foster, L.; Clapp, L.; Erickson, M.; Jabbari, B. Botulinum toxin A and chronic low back pain: A randomized, double-blind study. Neurology 2001, 56, 1290–1293. [Google Scholar] [CrossRef]
- Singh, J.A.; Mahowald, M.L.; Noorbaloochi, S. Intra-articular botulinum toxin A for refractory shoulder pain: A randomized, double-blinded, placebo-controlled trial. Transl. Res. 2009, 153, 205–216. [Google Scholar] [CrossRef]
- Singh, J.A.; Mahowald, M.L.; Noorbaloochi, S. Intraarticular botulinum toxin A for refractory painful total knee arthroplasty: A randomized controlled trial. J. Rheumatol. 2010, 37, 2377–2386. [Google Scholar] [CrossRef]
- McAlindon, T.E.; Schmidt, U.; Bugarin, D.; Abrams, S.; Geib, T.; DeGryse, R.E.; Kim, K.; Schnitzer, T.J. Efficacy and safety of single-dose onabotulinumtoxinA in the treatment of symptoms of osteoarthritis of the knee: Results of a placebo-controlled, double-blind study. Osteoarthr. Cartil. 2018, 26, 1291–1299. [Google Scholar] [CrossRef]
- Arendt-Nielsen, L.; Jiang, G.-L.; DeGryse, R.; Turkel, C.C. Intra-articular onabotulinumtoxinA in osteoarthritis knee pain: Effect on human mechanistic pain biomarkers and clinical pain. Scand. J. Rheumatol. 2017, 46, 303–316. [Google Scholar] [CrossRef]
- Safety and Efficacy Study of Botulinum Toxin Type A as Treatment for Osteoarthritis Knee Pain—Study Results—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01518257 (accessed on 4 July 2019).
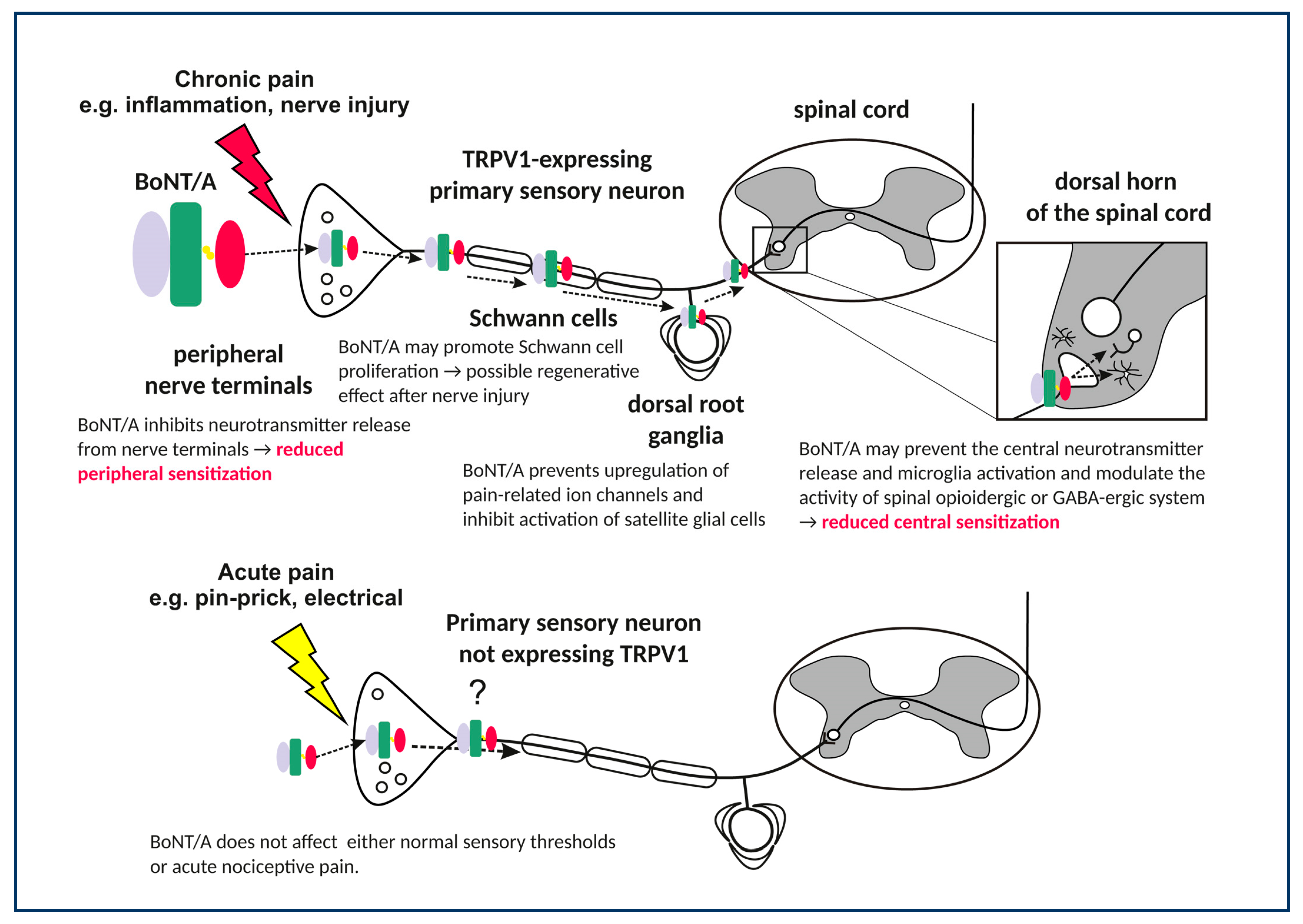

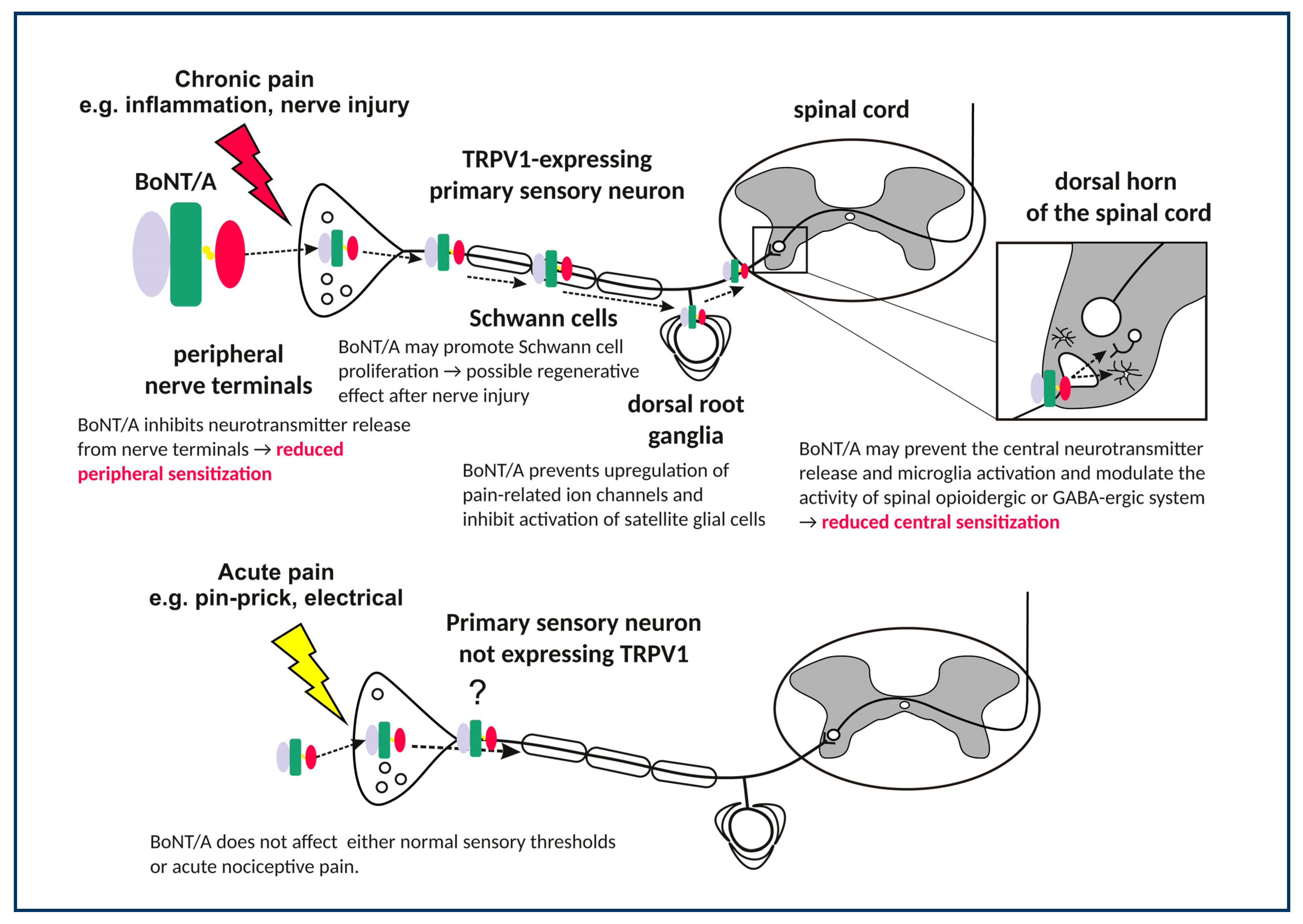{kind=link}
| General Factors not Specific to Pain | ||||
|---|---|---|---|---|
| Property of BoNT/A Molecule or Peculiarity of Action | Mechanisms of Action | Contribution to Desirable Pharmacological Properties | Attempted or Potential Improvement | References |
| Low local diffusion after application | rapid and high affinity binding to neuronal membrane at the injected site | safety, onset of the effect | lower injection volume, intradermal injections | [12,13,14] |
| Absorption through epithelial barriers | crossing epithelial barriers by transcytosis | application by different routes (e.g., transmucosal) | novel therapeutic systems with incorporated BoNT/A which might improve toxin absorption and extend the contact time with the epithelial tissue/mucosa | [15] |
| Specificity for hyperactive neurons | Expression of membrane acceptors such as glycosylated SV2C; higher rate of SVs exo/endocytosis favors toxin uptake | safety, selectivity for hyperactive nerve terminals | recombinant chimeras with different neuronal specificities | [16] |
| Protease specific targeting of SNARE proteins | synaptic localization, disturbance of SNARE supercomplex | potency, safety | - specific point mutations for higher affinity to SNAP-25 | [17] |
| - recombinant molecules with shorter action or different affinity for SNARE proteins | ||||
| Protease longevity inside neurons | cellular localization, avoidance of proteasomal degradation | long duration of effects | specific chimeras that change the affinity for intraneuronal degradation system | [18] |
| Reversibility of the neuroparalysis | recovery of neuronal exocytosis is dependent on nerve terminal type (gain of function) | long duration of effect (from 3 months to more than a year) | interference with the nerve function recovery processes | [19] |
| Repeatability of neuroparalysis | recovery of neuronal exocytosis can be repeated many times without loss of neuron function | application schedule | repeated application for prolonged period into the same site | [20] |
| Factors Specifically Related to Pain | ||||
| Property of BoNT/A Molecule or Peculiarity of Action | Mechanisms of Action | Contribution to Desirable Pharmacological Properties | Attempted or Potential Improvement | References |
| Selectivity for certain sensory neuron populations | occurrence in TRPV1-expressing neurons | selectivity for chronic or prolonged pain | recombinant chymeras with different receptor specificities | [21,22,23] |
| effect on glutamatergic transmission | efficacy in chronic pain (possibly LTP related) | |||
| effect on peptidergic transmitters | efficacy in chronic pain and migraine | |||
| Segmental activity in the sensory nucleus/spinal cord dorsal horn | microtubule-dependent neuronal axonal transport | localization of toxin effect | neural block for segmental treatment | [24,25,26] |
| Interaction with other pain neurotransmitter system | Interaction with endogenous opioid system | synergism with opioid analgesic and avoidance of tolerance development; efficacy in opioid-overused patients | combined use of lower dose opioids and BoNT/A | [27,28,29] |
| Restoration of sensitivity to morphine | ||||
| Model | BoNT (Type; Dose, Application) | Findings/Comments | Ref. |
|---|---|---|---|
| partial sciatic nerve injury in rats | A; 7 U/kg; i.pl.; injected after established hypersensitivity (day 14) | Long-term reduction of mechanical and thermal hypersensitivity (from day 5 after injection). First study on experimental peripheral neuropathic pain. | [45] |
| ligation of L5/L6 spinal nerve in rats | A; 10, 20, 30 or 40 U/kg i.pl. after established hypersenitivity | Reduction of mechanical allodynia (after 1 day) and cold allodynia (three days after injection; both effects lasted for 15 days after injection). The effect was dose-dependent. However, large systemic doses were used. | [80] |
| chronic constriction injury of the sciatic nerve in mice | A; 15 pg/mouse; i.pl. pre- and post-injury | Reduced mechanical allodynia (from day 1 after injection; lasting at least three weeks). BoNT/A reduced pain symptoms only if injected after neuropathy onset, but not as a pretreatment. | [81] |
| paclitaxel-induced peripheral polyneuropathy in rats | abobotulinumtoxinA (AboA); 20-30 U/kg; i.pl; post-treatment | Antihyperalgesic effect at both ipsilateral and contralateral paws (three and six days after injection). | [82] |
| streptozotocin diabetic neuropahy in rats | A; 3, 5 and 7 U/kg (i.pl); 1 U/kg (i.t.); post-treatment | Unilateral toxin application reduced mechanical and thermal hypersensitivity bilaterally (from fifth to 15th day after BoNT/A). Intrathecal BoNT-A was effective after 24h. Different onset and lower analgesic dose after intrathecal injection suggested central action of BoNT/A. | [83] |
| chronic constriction injury to the sciatic nerve in mice and in rats | A;1.875, 3.75, 7.5 and 15 pg/paw for mice; 18. 75 or 75 pg/paw or i.t. for rats; post-treatment on day 5 | Single i.pl. or i.t. injection significantly reduced the mechanical allodynia in mice and rats and thermal hyperalgesia in rats (from 24 h after toxin injection) and lasted for several weeks). Acceleration of regenerative processes in the injured nerve was also observed. | [84] |
| chronic constriction injury of the sciatic nerve in rats | A; 75 pg/paw; i.pl.; 3 days before and 5 days after CCI | Reduced neuropathic pain-related behavior and attenuated upregulation of NOS1, prodynorphin, pronociceptin mRNA in the DRG and microglia activation in both the spinal cord and DRG. | [85] |
| L5 ventral root transection (VRT) in rats | A; 7 U/kg; i.pl.; post-injury 4, 8 or 16 days | Reduced mechanical allodynia bilaterally and inhibited P2X (3) over-expression in DRG nociceptive neurons unulaterally to L5 VRT. | [46] |
| Infraorbital nerve constriction (IoNC) in rats | A; 3.5 U/kg into vibrissal pad; post-injury on day 14 | Unilateral toxin injection reduced the IoNC-induced dural extravasation and allodynia bilaterally (from day 2 and lasting 17 days after BoNT/A, prior to neuropathy resolution). Intraganglionic block of axonal transport by colchicine abolished the effects of BoNT/A. Bilateral effects of BoNT/A and dependence on retrograde axonal transport suggest a central site of its action. | [86] |
| transection of the L5 ventral root in rats | A; 10 or 20 U/kg, i.pl. post-injury at day 3 | Bilaterally decreased mechanical hyperalgesia, (from day 5, lasting at least 20 days post-BoNT/A). BoNT/A lowered the VRT-induced increased percentage of TRPV1 (+) neurons in the ipsilateral DRG. | [87] |
| chronic constriction injury in mice | A; 15 pg/paw i.pl.; post-injury at day 4 | Counteracted allodynia and reduced astrocyte activation. It increased the analgesic effect of morphine and countered morphine-induced tolerance. In neurons BoNT/A restored the expression of MORs reduced by repeated morphine administration. | [88] |
| partial sciatic nerve transection in rats | A; 7 U/kg, i.pl. post-injury at day 14 | Decreased mechanical and cold allodynia. Opioid antagonist naltrexone applied five days after the toxin reversed its antinociceptive effect. Central antinociceptive action of BoNT/A might be associated with the activity of endogenous opioid system via μ-opioid receptor. | [27] |
| partial sciatic nerve transection in rats | A; 7 U/kg, i.pl. post-injury at day 14 | Reduced mechanical allodynia. GABA-A antagonist bicuculine abolished the antinociceptive effect in toxin-treated animals, thus indicating involvement of central GABAergic system. | [89] |
| chronic constriction injury of the infraorbital nerve in rats | A; 3 or 10 U/kg; s.c. into the whisker pad; post-injuryt at day 14 | The toxin exerted antinociceptive effect and significantly lowered the expression of TRPA1, TRPV1, and TRPV2 in trigeminal nucleus caudalis (Vc); these effects were blocked by colchicine. | [90] |
| surgical constriction of the infraorbital nerve in rats | A; 15 U/kg; post-surgery at day 6; injected into the area of nerve ligation | Reduced thermal nociceptive response (TNR) beginning 6 h and lasting 72 h after treatment in senzitized animals. BoNT/A in sham group increased TNR thus suggesting a pronociceptive effect in non-sensitized animals. | [91] |
| malpositioned dental implants to induce injury to the inferior alveolar nerve in rats | A; 1 or 3 U/kg s.c. into the facial region; 3 days post-injury | Attenuated mechanical allodynia. Double treatments with 1 U/kg of BoNT-A produced prolonged, more antiallodynic effects as compared with single treatments. BoNT-A significantly inhibited the upregulation of Nav1.7 expression in the trigeminal ganglion in the nerve-injured animals. | [92] |
| chronic constriction injury of the sciatic nerve in rats | A; 300 pg/paw; i.pl. post-surgery at day 5 | Attenuated pain-related behavior and microglial activation. It restored the neuroimmune balance by decreasing the levels of pronociceptive factors (IL-1β and IL-18) and increasing the levels of antinociceptive factors (IL-10 and IL-1RA) in the spinal cord and DRG. | [93] |
| streptozotocin-induced diabetic polyneuropathy; chronic constriction injury in rats | aboA; 15 or 20 U/kg; s.c.; post-injection and post-injury at day 14 | Unilateral aboA reduced bilateral mechanical hyperalgesia in diabetic polyneuropathy model, while had no effect on unilateral CCI-induced hyperalgesia if applied contralaterally to the injury. | [94] |
| rat spared nerve injury (SNI) model | LC/E-BoNT/A chimera; 15–75 U/kg, i.pl. post-surgery at day 4 | Alleviated for ∼two weeks mechanical and cold hyper-sensitivities. When injected five weeks after injury, LC/E-BoNT/A still reversed fully-established mechanical and cold hyper-sensitivity. | [18] |
| partial sciatic nerve ligation in mice (SP and NK1R knockout mice) | A; 0.2 and 0.4 U/paw, i.pl. post-surgery at day 7 | Reduced hyperalgesia in wild type animals, but not in gene-deleted groups, suggesting the necessary involvement of SP-ergic system in the antinociceptive activity of BoNT/A. | [95] |
| Pain Condition | Number of Participants | Dose and Delivery Route | Primary Outcome | Reference | |
|---|---|---|---|---|---|
| posttraumatic neuralgia 1 | 29 | 5 U/site max. 200 U i.d. | pain rating 0–10 | BoNT/A − 1.9 placebo − 0.3 | [34] |
| posttraumatic neuralgia 2 | 46 | 5 U/site max. 300 U i.d. | pain rating 0–10 | BoNT/A − 1.9 placebo − 0.6 | [107] |
| postherpetic neuralgia | 60 | 5 U/site max. 200 U s.c. | pain rating 0–10 | BoNT/A − 4.5 lidocaine − 2.6 placebo − 2.9 | [141] |
| postherpetic neuralgia | 30 | 5 U/site max. 100 U s.c. | pain rating 0–10 | BoNT/A − 4.6 placebo − 0.5 | [142] |
| postherpetic neuralgia | 117 | 2.5 U/site max. 200 U i.d. | pain rating 0–10 | BoNT/A − 1.2 placebo − 1.2 | [140] |
| trigeminal neuralgia | 42 | 5 U/site, 75 U i.d. or s.m. | pain rating 0–10 | BoNT/A − 6.05 placebo − 1.88 | [143] |
| trigeminal neuralgia | 20 | 5U/site, 100 U s.c. | pain rating (0–10) frequency of paroxysms/day | BoNT/A − 6.5 placebo − 0.3 BoNT/A − 32.7 placebo − 0.1 | [144] |
| trigeminal neuralgia | 36 | 50 U s.c. | pain rating (0–10) frequency of paroxysms/day | BoNT/A − 4.1 placebo − 1.25 BoNT/A − 22.0 placebo − 9.81 | [145] |
| trigeminal neuralgia | 80 | 20 sites 25 or 75U i.d. or s.m. | pain rating 0–10 | BoNT/A 25U − 4.24 BoNT/A 75U − 5.4 placebo − 2.96 | [146] |
| diabetic neuropathy | 18 | 4U/site, 50U | pain rating 0–10 | BoNT/A − 2.53 placebo − 0.53 | [147] |
| Pain Condition | Number of Participants | Dose and Delivery Route | Primary Outcome | Reference | |
|---|---|---|---|---|---|
| low back pain | 31 | 40 U/site 200 U i.m. | % of responders (50% reduction in pain rating) | BoNT/A 73.3% placebo 25% | [149] |
| refractory shoulder pain | 36 | 100 U i.a. | pain rating 0–10 | BoNT/A -2.4 placebo -0.8 | [150] |
| refractory painful total knee arthroplasty | 54 | 100 U i.a. | % of responders (2-point reduction in pain ratings) | BoNT/A 71% placebo 35% | [151] |
| knee osteoarthritis | 176 | 200 U or 400 U i.a. | pain rating 0–10 | BoNT/A 200 U -1.6 BoNT/A 400U -2.1 placebo -2.1 | [152] |
| knee osteoarthritis | 121 | 200 U i.a. | pain rating 0–10 | BoNT/A -2.2 placebo -2.5 | [153,154] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matak, I.; Bölcskei, K.; Bach-Rojecky, L.; Helyes, Z. Mechanisms of Botulinum Toxin Type A Action on Pain. Toxins 2019, 11, 459. https://doi.org/10.3390/toxins11080459
Matak I, Bölcskei K, Bach-Rojecky L, Helyes Z. Mechanisms of Botulinum Toxin Type A Action on Pain. Toxins. 2019; 11(8):459. https://doi.org/10.3390/toxins11080459
Chicago/Turabian StyleMatak, Ivica, Kata Bölcskei, Lidija Bach-Rojecky, and Zsuzsanna Helyes. 2019. "Mechanisms of Botulinum Toxin Type A Action on Pain" Toxins 11, no. 8: 459. https://doi.org/10.3390/toxins11080459
APA StyleMatak, I., Bölcskei, K., Bach-Rojecky, L., & Helyes, Z. (2019). Mechanisms of Botulinum Toxin Type A Action on Pain. Toxins, 11(8), 459. https://doi.org/10.3390/toxins11080459