Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
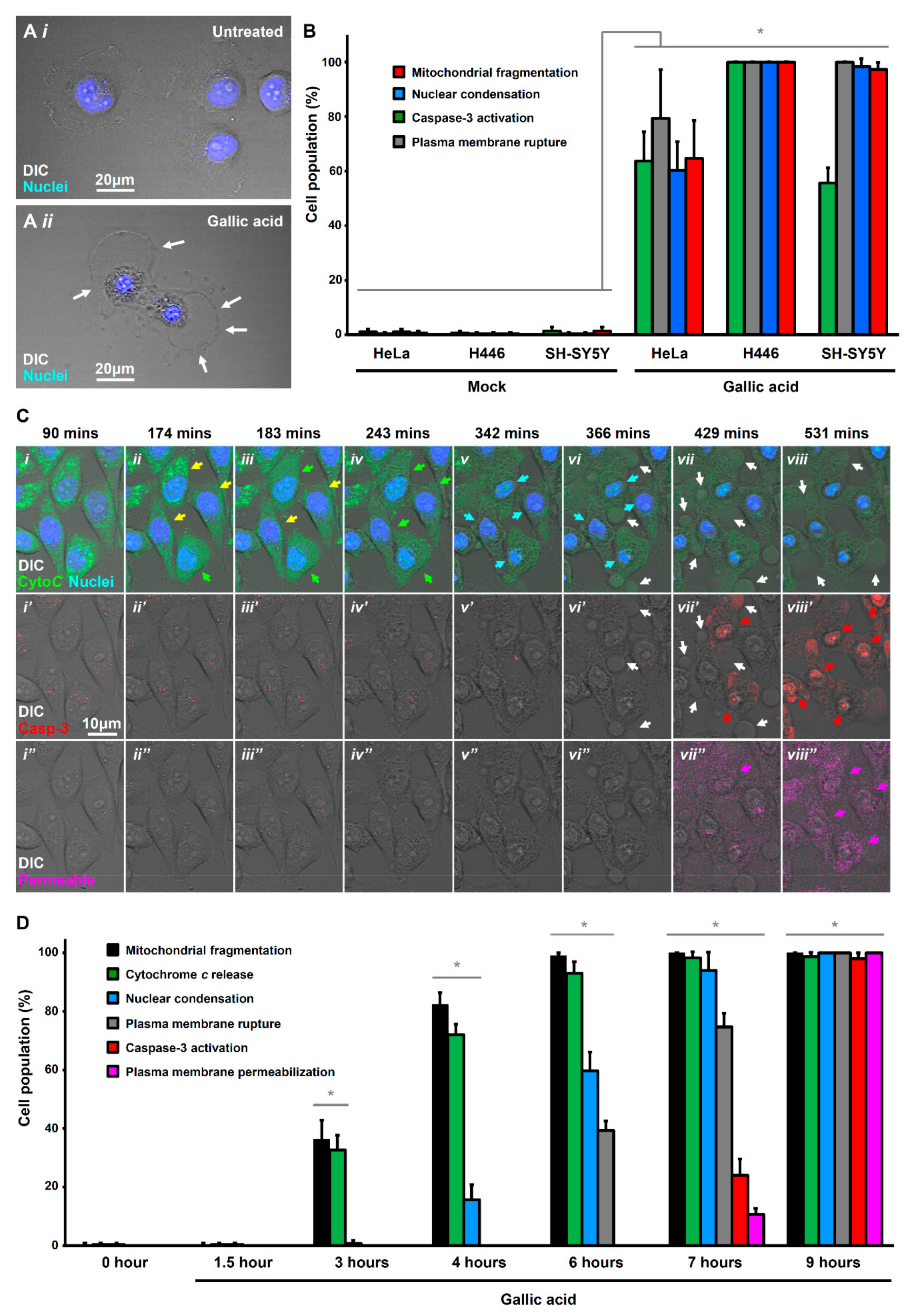
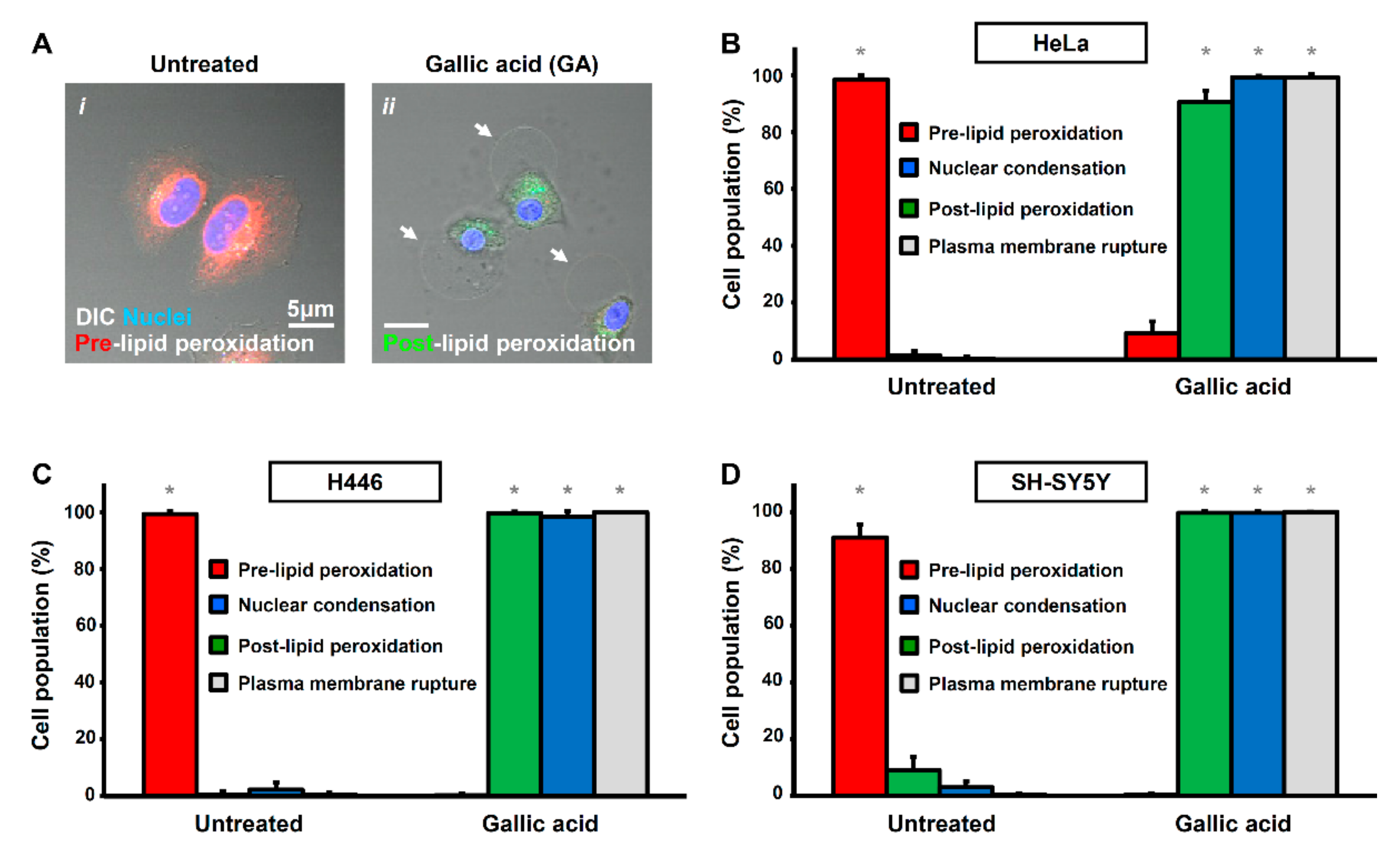
2.1. Activation of Apoptotic Cell Death by Gallic Acid
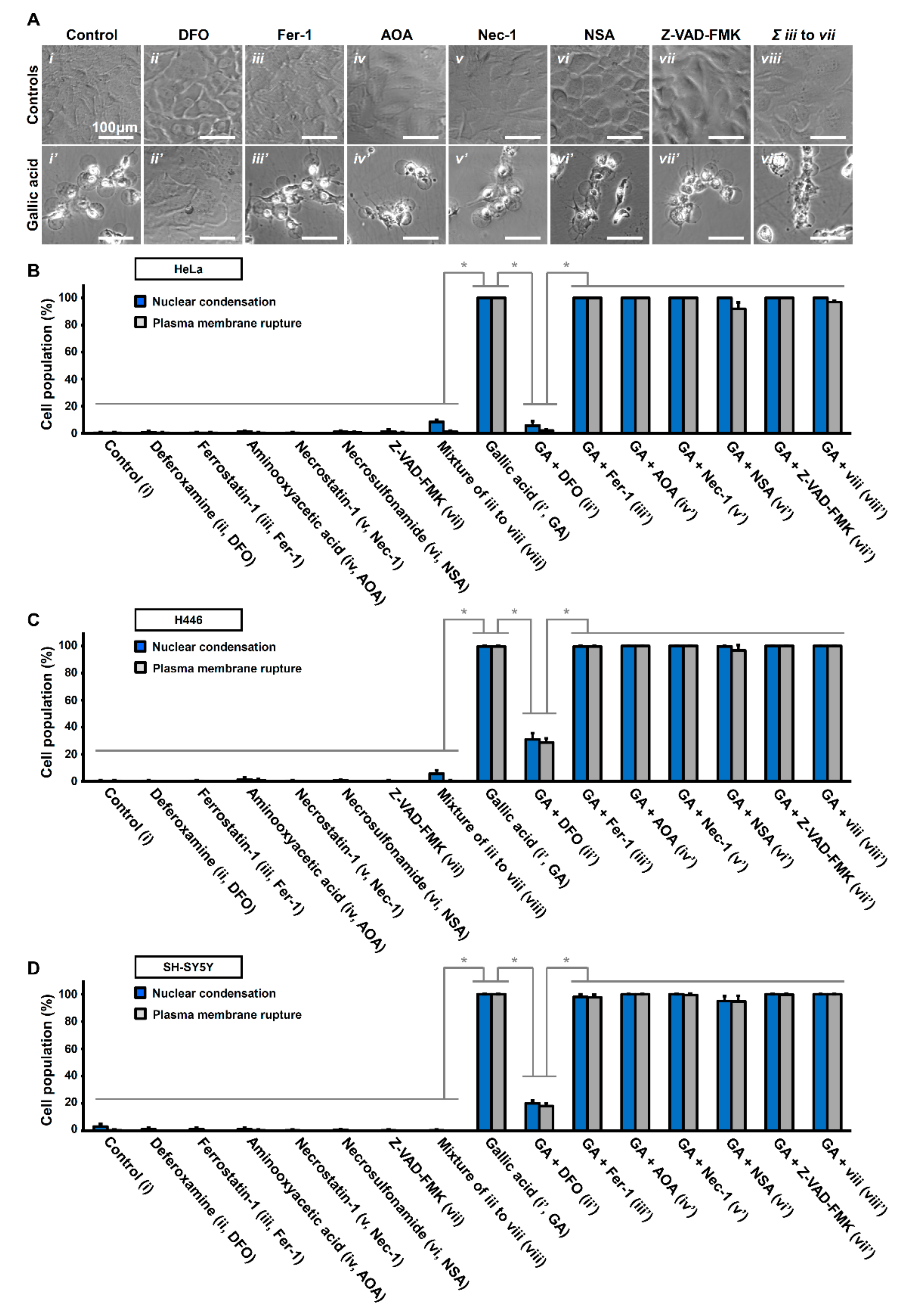
2.2. Activation of Necroptotic Pathway by Gallic Acid
2.3. Activation of Ferroptotic Pathway by Gallic Acid
2.4. Other Unidentified Cell Death Mechanisms Triggered by Gallic Acid
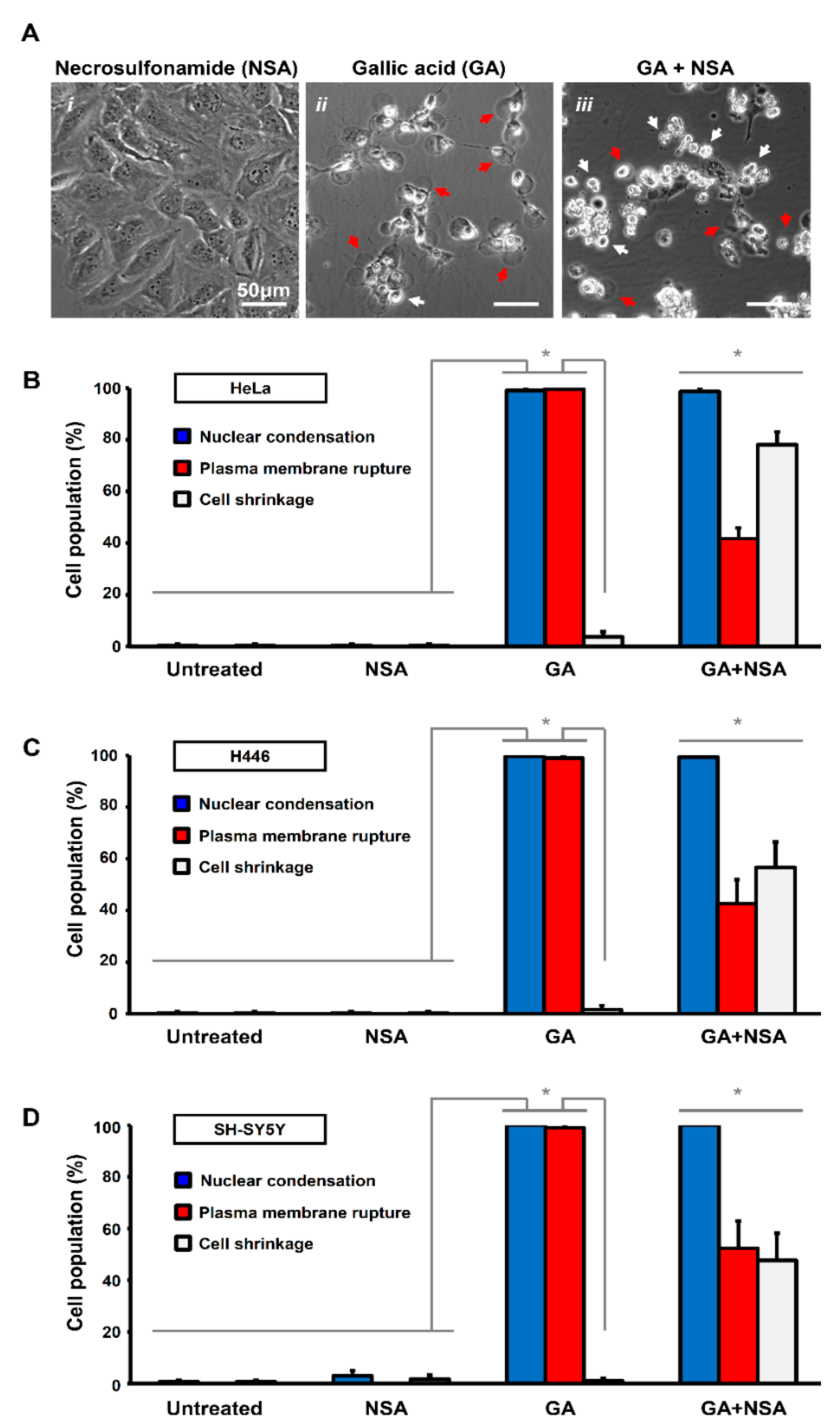
2.5. Synergistic Effect of Necrosulfonamide and Gallic Acid in Cell Death Induction
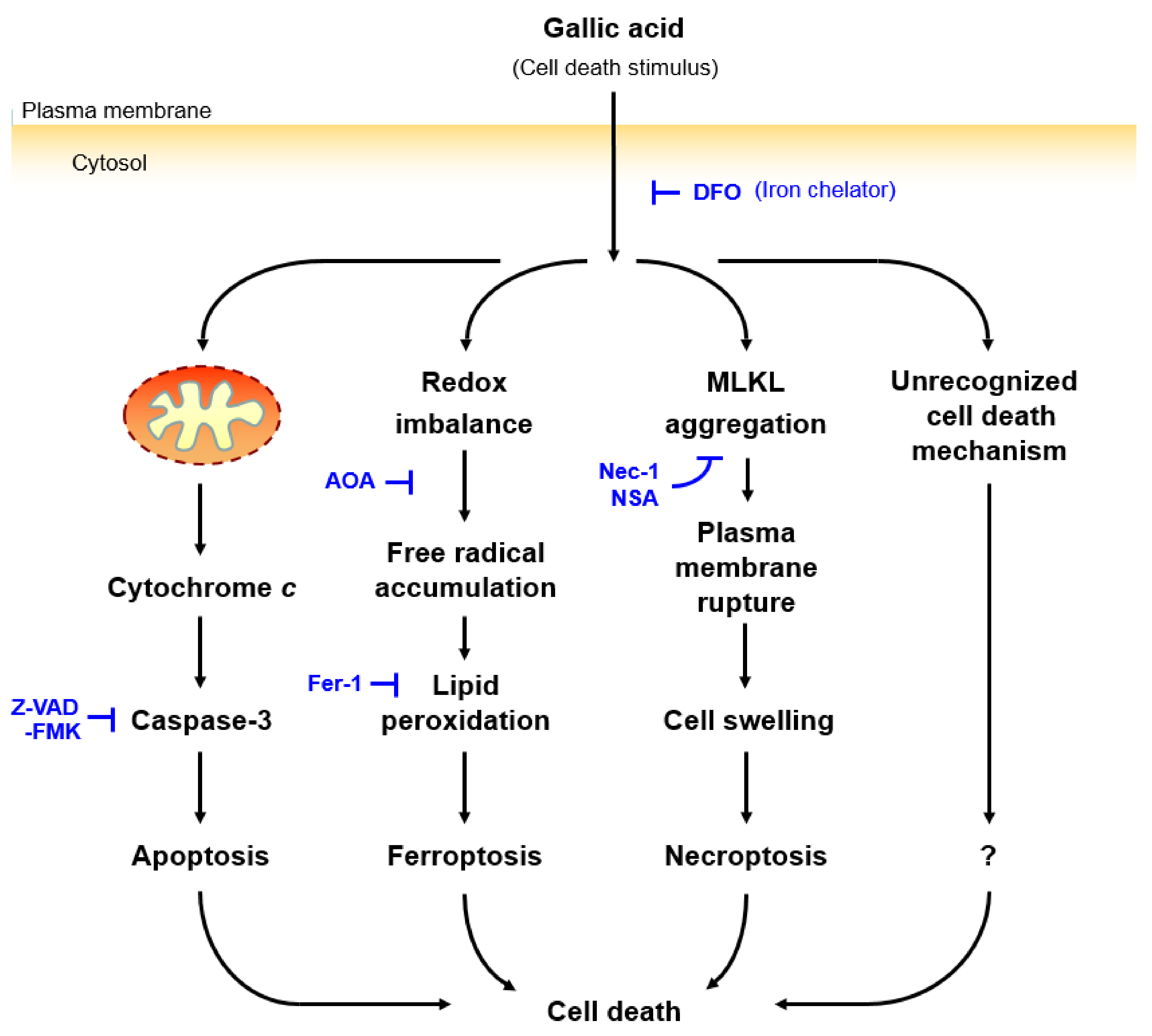
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Death Induction
4.4. Live Cell Staining
4.5. Confocal Microscopy
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aminooxyacetic acid | AOA |
| Cytochrome c | CytoC |
| Deferoxamine | DFO |
| Differential Interference Contrast | DIC |
| Dimethyl Sulfoxide | DMSO |
| Ferrostatin-1 | Fer-1 |
| Gallic acid | GA |
| Low Molecular Weight | LMW |
| Necrostatin-1 | Nec-1 |
| Necrosulfonamide | NSA |
References
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Brown, J.M.; Attardi, L.D. The role of apoptosis in cancer development and treatment response. Nat. Rev. Cancer 2005, 5, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Demedts, I.K.; Vermaelen, K.Y.; van Meerbeeck, J.P. Treatment of extensive-stage small cell lung carcinoma: Current status and future prospects. Eur. Respir. J. 2010, 35, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Ewer, M.S.; Ewer, S.M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 2015, 12, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M. Preventing or reducing late side effects of radiation therapy: Radiobiology meets molecular pathology. Nat. Rev. Cancer 2006, 6, 702–713. [Google Scholar] [CrossRef]
- Ramirez, L.Y.; Huestis, S.E.; Yap, T.Y.; Zyzanski, S.; Drotar, D.; Kodish, E. Potential chemotherapy side effects: What do oncologists tell parents? Pediatr. Blood Cancer 2009, 52, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Tannock, I.F. Repopulation of cancer cells during therapy: An important cause of treatment failure. Nat. Rev. Cancer 2005, 5, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Tannock, J.F. Repopulation of tumour cells between cycles of chemotherapy: A neglected factor. Lancet Oncol. 2000, 1, 86–93. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, S.W.; Cheung, P.C.K.; Wang, Q. Antitumor polysaccharides from mushrooms: A review on their isolation process, structural characteristics and antitumor activity. Trends Food Sci. Tech. 2006, 18, 4–19. [Google Scholar] [CrossRef]
- Sanders, K.; Moran, Z.; Shi, Z.; Paul, R.; Greenlee, H. Natural Products for Cancer Prevention: Clinical Update 2016. Semin. Oncol. Nurs. 2016, 32, 215–240. [Google Scholar] [CrossRef] [PubMed]
- Valverde, M.E.; Hernandez-Perez, T.; Paredes-Lopez, O. Edible mushrooms: Improving human health and promoting quality life. Int. J. Microbiol. 2015, 2015, 376387. [Google Scholar] [CrossRef] [PubMed]
- Wasser, S.P. Medicinal mushroom science: Current perspectives, advances, evidences, and challenges. Biomed. J. 2014, 37, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Goyal, A. Recent developments in mushrooms as anti-cancer therapeutics: A review. 3 Biotech 2012, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.; Evidente, A.; Vurro, M.; Mathieu, V.; Cimmino, A.; Evidente, M.; van Otterlo, W.A.; Dasari, R.; Lefranc, F.; Kiss, R. Toward a Cancer Drug of Fungal Origin. Med. Res. Rev. 2015, 35, 937–967. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Lai, T.C.; Lam, K.L.; Cheung, P.C.K. Anti-angiogenic effect of water extract from the fruiting body of Agrocybe aegerita. LWT Food Sci. Technol. 2017, 75, 155–163. [Google Scholar] [CrossRef]
- Ferreira, I.C.; Vaz, J.A.; Vasconcelos, M.H.; Martins, A. Compounds from wild mushrooms with antitumor potential. Anticancer Agents Med. Chem. 2010, 10, 424–436. [Google Scholar] [CrossRef]
- Lin, S.; Ching, L.T.; Ke, X.; Cheung, P.C. Comparison of the Composition and Antioxidant Activities of Phenolics from the Fruiting Bodies of Cultivated Asian Culinary-Medicinal Mushrooms. Int. J. Med. Mushrooms 2016, 18, 871–881. [Google Scholar] [CrossRef]
- Subramanian, A.P.; John, A.A.; Vellayappan, M.V.; Balaji, A.; Jaganathan, S.K.; Supriyanto, E.; Yusof, M. Gallic acid: Prospects and molecular mechanisms of its anticancer activity. Rsc. Adv. 2015, 5, 35608–35621. [Google Scholar] [CrossRef]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Aglan, H.A.; Ahmed, H.H.; El-Toumy, S.A.; Mahmoud, N.S. Gallic acid against hepatocellular carcinoma: An integrated scheme of the potential mechanisms of action from in vivo study. Tumour Biol. 2017, 39, 1010428317699127. [Google Scholar] [CrossRef]
- Vijaya Padma, V.; Sowmya, P.; Arun Felix, T.; Baskaran, R.; Poornima, P. Protective effect of gallic acid against lindane induced toxicity in experimental rats. Food Chem. Toxicol. 2011, 49, 991–998. [Google Scholar] [CrossRef]
- Chuang, C.Y.; Liu, H.C.; Wu, L.C.; Chen, C.Y.; Chang, J.T.; Hsu, S.L. Gallic acid induces apoptosis of lung fibroblasts via a reactive oxygen species-dependent ataxia telangiectasia mutated-p53 activation pathway. J. Agric. Food Chem. 2010, 58, 2943–2951. [Google Scholar] [CrossRef]
- Aborehab, N.M.; Osama, N. Effect of Gallic acid in potentiating chemotherapeutic effect of Paclitaxel in HeLa cervical cancer cells. Cancer Cell Int. 2019, 19, 154. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]
- Silva, M.T. Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Lett. 2010, 584, 4491–4499. [Google Scholar] [CrossRef] [Green Version]
- Strzyz, P. Cell death: Pulling the apoptotic trigger for necrosis. Nat. Rev. Mol. Cell Biol. 2017, 18, 72. [Google Scholar] [CrossRef]
- Goldstein, J.C.; Waterhouse, N.J.; Juin, P.; Evan, G.I.; Green, D.R. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2000, 2, 156–162. [Google Scholar] [CrossRef]
- Goldstein, J.C.; Munoz-Pinedo, C.; Ricci, J.E.; Adams, S.R.; Kelekar, A.; Schuler, M.; Tsien, R.Y.; Green, D.R. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005, 12, 453–462. [Google Scholar] [CrossRef]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar]
- Bhola, P.D.; Letai, A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Canc. Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.M.; Cheung, P.C.K. Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins 2019, 11, 492. https://doi.org/10.3390/toxins11090492
Tang HM, Cheung PCK. Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins. 2019; 11(9):492. https://doi.org/10.3390/toxins11090492
Chicago/Turabian StyleTang, Ho Man, and Peter Chi Keung Cheung. 2019. "Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features" Toxins 11, no. 9: 492. https://doi.org/10.3390/toxins11090492
APA StyleTang, H. M., & Cheung, P. C. K. (2019). Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins, 11(9), 492. https://doi.org/10.3390/toxins11090492