Inhibition of Pore-Forming Proteins


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Toxic Pore-Forming Proteins
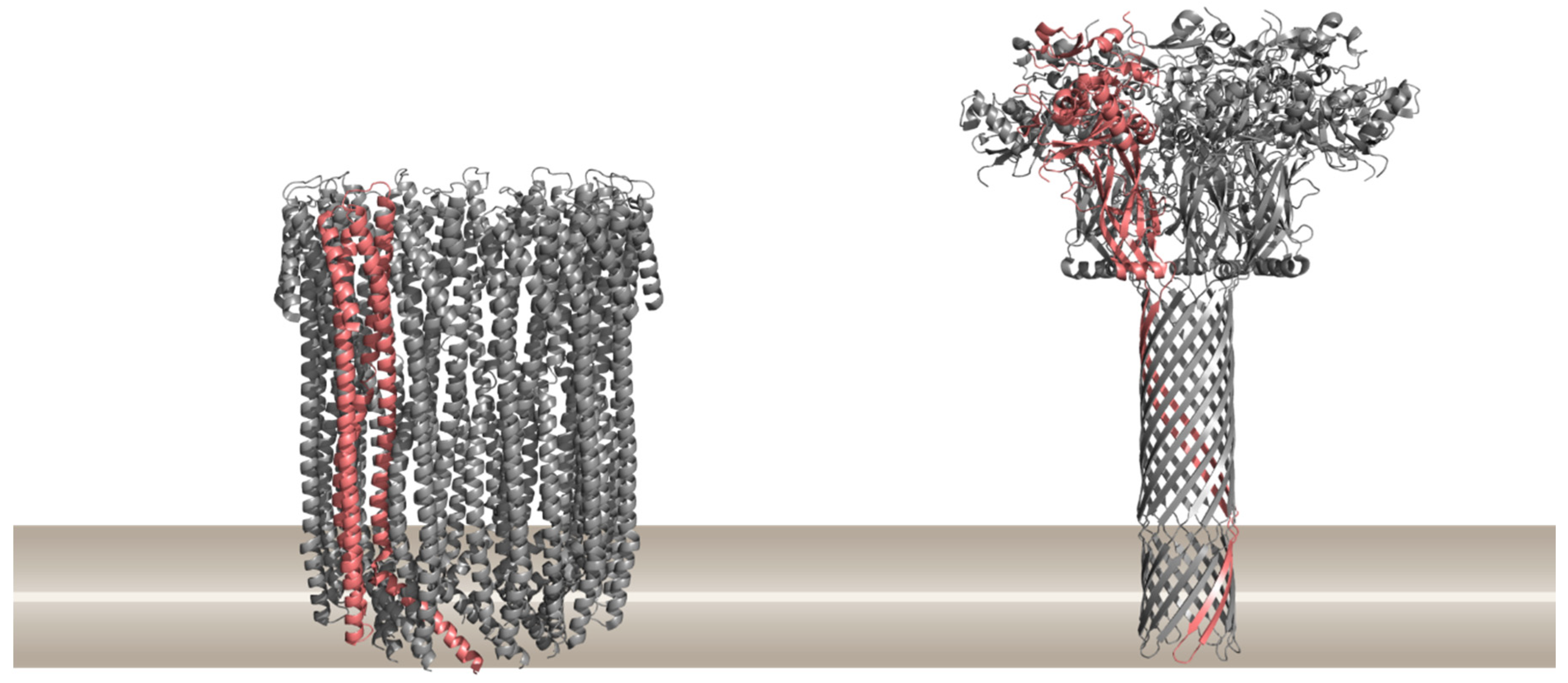
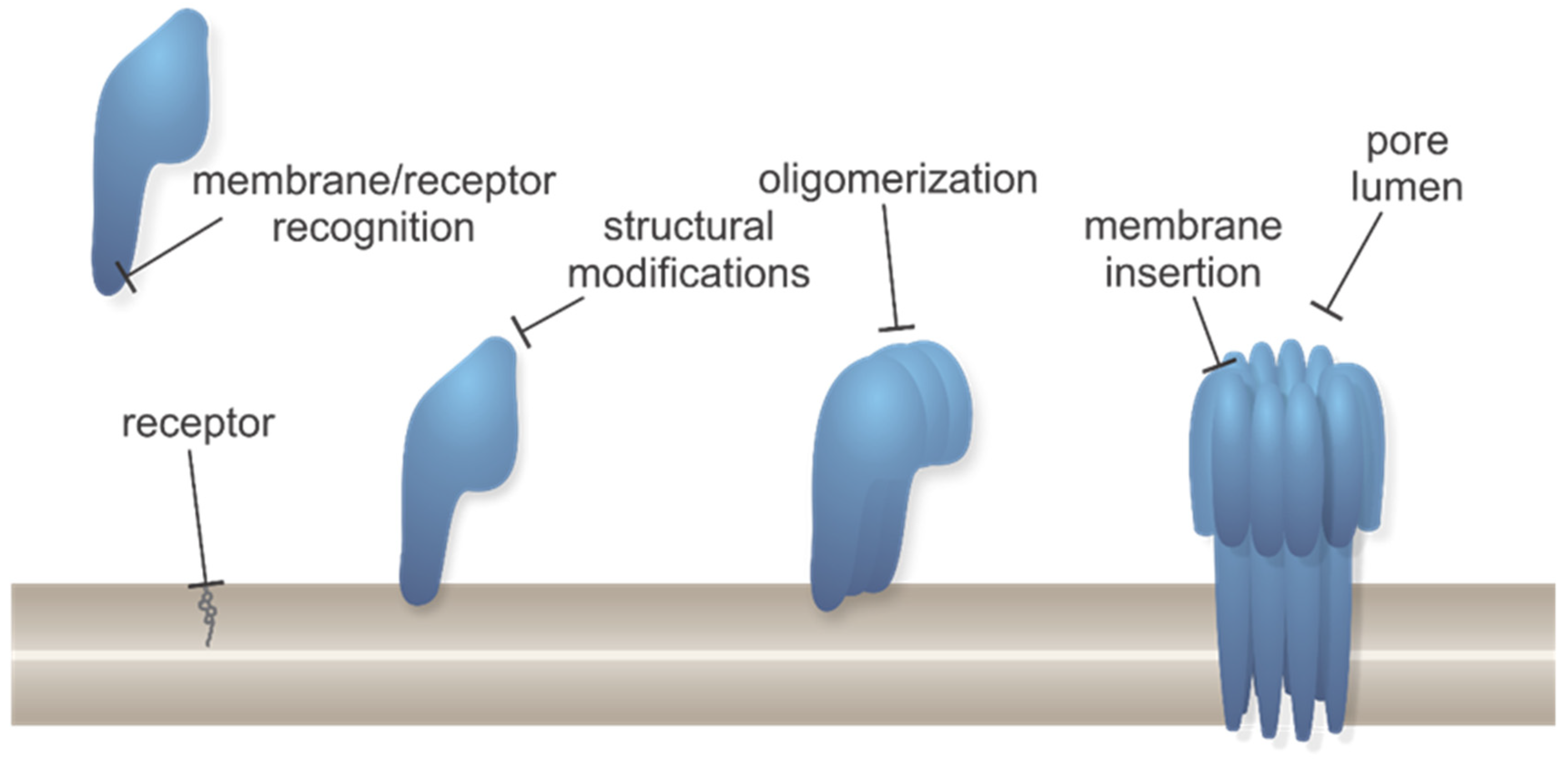
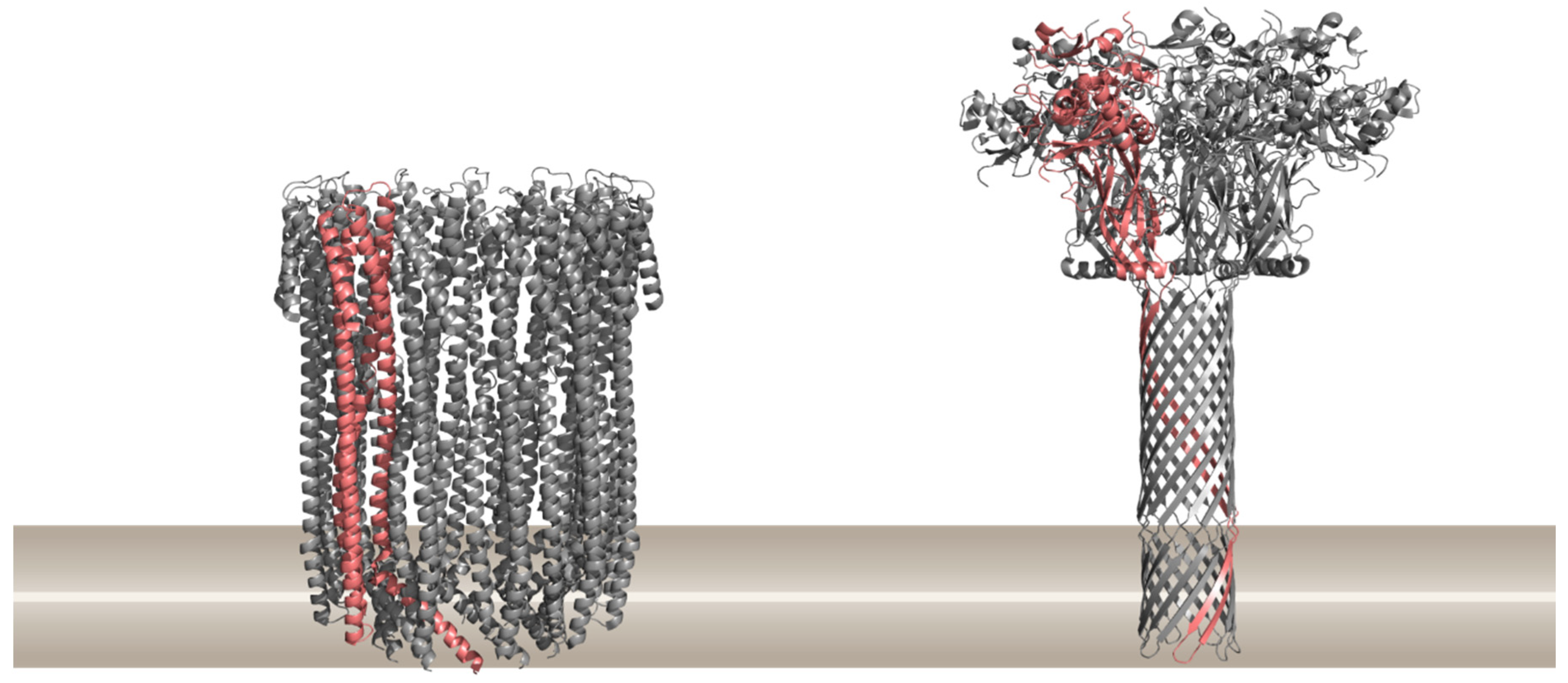
1.1. Different Modes of Creating a Pore in Cellular Membranes
1.2. Effects of PFPs on Target Cells and Their Biological Roles
2. Modes of Preventing Pore Formation
2.1. Small Molecules
2.2. Synthetic Nanoparticles
2.3. Neutralizing Antibodies
2.4. Antibody-Derived Scaffolds and Antibody Mimetics
2.5. Polyvalent Inhibitors
2.6. Receptor-Like Decoys for Pore-Forming Toxins
2.7. Dominant Negative Mutants
3. Conclusions
Funding
Conflicts of Interest
References
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic pore-forming proteins: Function and host response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Lakey, J.H. Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 2008, 33, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.J.C.; Dalla Serra, M.; Froelich, C.J.; Wallace, M.I.; Anderluh, G. Membrane pore formation at protein-lipid interfaces. Trends Biochem. Sci. 2014, 39, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Cajnko, M.M.; Mikelj, M.; Turk, T.; Podobnik, M.; Anderluh, G. Membrane Interactions and Cellular Effects of MACPF/CDC Proteins. In MACPF/CDC Proteins—Agents of Defence, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 119–144. [Google Scholar]
- Lakey, J.H.; Anderluh, G. Membrane-Disrupting Proteins. In Biogenesis of Fatty Acids, Lipids and Membranes; Springer International Publishing: Dordrecht, The Netherlands, 2019; pp. 729–739. [Google Scholar]
- Ros, U.; García-Sáez, A.J. More Than a Pore: The Interplay of Pore-Forming Proteins and Lipid Membranes. J. Membr. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dunstone, M.A.; Tweten, R.K. Packing a punch: The mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr. Opin. Struct. Biol. 2012, 22, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Tweten, R.K.; Hotze, E.M.; Wade, K.R. The unique molecular choreography of giant pore formation by the cholesterol-dependent cytolysins of Gram-positive bacteria. Annu. Rev. Microbiol. 2015, 69, 323–340. [Google Scholar] [CrossRef]
- Krasilnikov, O.V.; Da Cruz, J.B.; Yuldasheva, L.N.; Varanda, W.A.; Nogueira, R.A. A novel approach to study the geometry of the water lumen of ion channels: Colicin Ia channels in planar lipid bilayers. J. Membr. Biol. 1998, 161, 83–92. [Google Scholar] [CrossRef]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Cosentino, K.; Ros, U.; García-Sáez, A.J. Assembling the puzzle: Oligomerization of α-pore forming proteins in membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 457–466. [Google Scholar] [CrossRef]
- Gouaux, E. Channel-forming toxins: Tales of transformation. Curr. Opin. Struct. Biol. 1997, 7, 566–573. [Google Scholar] [CrossRef]
- Podobnik, M.; Savory, P.; Rojko, N.; Kisovec, M.; Wood, N.; Hambley, R.; Pugh, J.; Wallace, E.J.; McNeill, L.; Bruce, M.; et al. Crystal structure of an invertebrate cytolysin pore reveals unique properties and mechanism of assembly. Nat. Commun. 2016, 7, 11598. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nagahama, M.; Hisatsune, J.; Katunuma, N.; Tsuge, H. Clostridium perfringens ι-toxin, ADP-ribosyltransferase: Structure and mechanism of action. Adv. Enzym. Regul. 2003, 43, 361–377. [Google Scholar] [CrossRef]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pentelute, B.L.; Collier, R.J.; Hong Zhou, Z. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 2015, 521, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Serna, M.; Giles, J.L.; Morgan, B.P.; Bubeck, D. Structural basis of complement membrane attack complex formation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.E.; Wong, T.Y.; Schwarzenbacher, R.; Jarrell, E.T.; Leppla, S.H.; Collier, R.J.; Liddington, R.C.; Cantley, L.C. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 2004, 11, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Dunstone, M.A.; Baran, K.; Whisstock, J.C.; Trapani, J.A. Perforin: Structure, function, and role in human immunopathology. Immunol. Rev. 2010, 235, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Moayeri, M.; Leppla, S.H.; Bezrukov, S.M. Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc. Natl. Acad. Sci. USA 2005, 102, 15075–15080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.; Kate, S.; Poon, V.; Mondal, D.; Boggara, M.B.; Saraph, A.; Martin, J.T.; McAlpine, R.; Day, R.; Garcia, A.E.; et al. Structure-based design of a heptavalent anthrax toxin inhibitor. Biomacromolecules 2011, 12, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.P.; Johnstone, B.A.; Tweten, R.K.; Parker, M.W.; Morton, C.J. Cholesterol-dependent cytolysins: From water-soluble state to membrane pore. Biophys. Rev. 2018, 10, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Rojko, N.; Dalla Serra, M.; Maček, P.; Anderluh, G. Pore formation by actinoporins, cytolysins from sea anemones. Biochim. Biophys. Acta Biomembr. 2016, 1858, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Dalla Serra, M.; Viero, G.; Guella, G.; Maček, P.; Menestrina, G. Pore formation by equinatoxin II, a eukaryotic protein toxin, occurs by induction of nonlamellar lipid structures. J. Biol. Chem. 2003, 278, 45216–45223. [Google Scholar] [CrossRef] [PubMed]
- Sobko, A.A.; Kotova, E.A.; Antonenko, Y.N.; Zakharov, S.D.; Cramer, W.A. Effect of lipids with different spontaneous curvature on the channel activity of colicin E1: Evidence in favor of a toroidal pore. FEBS Lett. 2004, 576, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Martinou, J.C.; Montessuit, S.; Epand, R.M.; Yip, C.M. Direct evidence for membrane pore formation by the apoptotic protein Bax. Biochem. Biophys. Res. Commun. 2002, 298, 744–749. [Google Scholar] [CrossRef]
- Henkel, J.S.; Baldwin, M.R.; Barbieri, J.T. Toxins from bacteria. In Molecular, Clinical and Environmental Toxicology. Experientia Supplementum; Luch, A., Ed.; Birkhäuser: Basel, Switzerland, 2010; Volume 100. [Google Scholar]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-Toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar]
- Popoff, M.R. Epsilon toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Barth, H. The actin-ADP-ribosylating Clostridium botulinum C2 toxin. Anaerobe 2004, 10, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Gilbert, R. MACPF/CDC Proteins—Agents of Defence, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer: Berlin, Germany, 2014. [Google Scholar]
- van der Goot, G. Pore Forming Toxins; Springer: Berlin, Germany, 2001. [Google Scholar]
- Bubeck Wardenburg, J.; Whisstock, J.; Tweten, R.K. Pore-forming toxins. In Bacterial Toxins: Genetics, Cellular Biology and Practical Applications; Proft, T., Ed.; Caister Academic Press: Wymondham, UK, 2013. [Google Scholar]
- Dalla Serra, M.; Teyuca Martinez, M. Pore-forming Toxins. In Encyclopedia of Life Sciences; John Wiley&Sons, Ltd.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Iacovache, I.; van der Goot, F.G.; Pernot, L. Pore formation: An ancient yet complex form of attack. Biochim. Biophys. Acta 2008, 1778, 1611–1623. [Google Scholar] [CrossRef] [Green Version]
- Los, F.C.O.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 9 September 2019).
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J.; Young, J.A.T. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.A.; Bradley, K.A.; Young, J.A.T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The Protective Antigen Component of Anthrax Toxin Forms Functional Octameric Complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef]
- Froude, J.W.; Thullier, P.; Pelat, T. Antibodies Against Anthrax: Mechanisms of Action and Clinical Applications. Toxins 2011, 3, 1433–1452. [Google Scholar] [CrossRef]
- Little, S.F. Anthrax vaccines: A development update. BioDrugs 2005, 19, 233–245. [Google Scholar] [CrossRef]
- Ambrose, E.A. Botulinum Neurotoxin, Tetanus Toxin, and Anthrax Lethal Factor Countermeasures. In Topics in Medicinal Chemistry; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Bouzianas, D.G. Current and future medical approaches to combat the anthrax threat. J. Med. Chem. 2010, 53, 4305–4331. [Google Scholar] [CrossRef]
- Laventie, B.; Potrich, C.; Atmanène, C.; Saleh, M.; Joubert, O.; Viero, G.; Bachmeyer, C.; Antonini, V.; Mancini, I.; Cianferani-Sanglier, S.; et al. p-Sulfonato-calix[n]arenes inhibit staphylococcal bicomponent leukotoxins by supramolecular interactions. Biochem. J. 2013, 450, 559–571. [Google Scholar] [CrossRef] [Green Version]
- LaRosa, S.P.; Opal, S.M. Sepsis Strategies in Development. Clin. Chest Med. 2008, 29, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lakey, J.H.; van der Goot, F.G.; Pattus, F. All in the family: The toxic activity of pore-forming colicins. Toxicology 1994, 87, 85–108. [Google Scholar] [CrossRef]
- Bullock, J.O.; Kolen, E.R.; Shear, J.L. Ion Selectivity of Colicin El: II. Permeability to Organic Cations. J. Membr. Biol. 1992, 128. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R. Molecular composition of the tubular structure of the membrane attack complex of complement. J. Biol. Chem. 1984, 259, 8641–8647. [Google Scholar] [PubMed]
- Thiery, J.; Keefe, D.; Boulant, S.; Boucrot, E.; Martinvalet, D.; Goping, I.S.; Bleackley, R.C.; Lieberman, J. Perforin pores in the endosomal membrane trigger release of endocytosed granzyme B to the cytosol of target cells. Nat. Immunol. 2011, 12, 770–777. [Google Scholar] [CrossRef]
- Gross, A.; Mcdonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 2001, 26, 61–66. [Google Scholar] [CrossRef]
- Nestorovich, E.M.; Bezrukov, S.M. Obstructing toxin pathways by targeted pore blockage. Chem. Rev. 2012, 112, 6388–6430. [Google Scholar] [CrossRef]
- Escajadillo, T.; Nizet, V. Pharmacological Targeting of Pore-Forming Toxins as Adjunctive Therapy for Invasive Bacterial Infection. Toxins 2018, 10. [Google Scholar] [CrossRef]
- Bezrukov, S.M.; Nestorovich, E.M. Inhibiting bacterial toxins by channel blockage. FEMS Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef]
- Hung, D.T.; Shakhnovich, E.A.; Pierson, E.; Mekalanos, J.J. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 2005, 310, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Hentzer, M.; Wu, H.; Andersen, J.B.; Riedel, K.; Rasmussen, T.B.; Bagge, N.; Kumar, N.; Schembri, M.A.; Song, Z.; Kristoffersen, P.; et al. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 2003, 22, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Statt, S.; Ruan, J.W.; Hung, L.Y.; Chang, C.Y.; Huang, C.T.; Lim, J.H.; Li, J.D.; Wu, R.; Kao, C.Y. Statin-conferred enhanced cellular resistance against bacterial pore-forming toxins in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 53, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Shewell, L.K.; Harvey, R.M.; Higgins, M.A.; Day, C.J.; Hartley-Tassell, L.E.; Chen, A.Y.; Gillen, C.M.; James, D.B.A.; Alonzo, F.; Torres, V.J.; et al. The cholesterol-dependent cytolysins pneumolysin and streptolysin O require binding to red blood cell glycans for hemolytic activity. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Hundhausen, C.; Lambert, M.; Broadway, N.; Andrews, R.; Bickett, D.; Leesnitzer, M.; Becherer, J. Metalloproteinase Inhibitors for the Disintegrin-Like Metalloproteinases ADAM10 and ADAM17 that Differentially Block Constitutive and Phorbol Ester-Inducible Shedding of Cell Surface Molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F.; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; Dumont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 Activation of Lipid Metabolic Pathways in Response to Bacterial Pore-Forming Toxins Promotes Cell Survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Yarovinsky, T.O.; Monick, M.M.; Husmann, M.; Hunninghake, G.W. Interferons increase cell resistance to staphylococcal alpha-toxin. Infect. Immun. 2008, 76, 571–577. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef]
- Cunha, B.A. Antibiotic Side Effects. Med. Clin. N. Am. 2001, 85, 149–185. [Google Scholar] [CrossRef]
- Bromberg-White, J.L.; Duesbery, N.S. Biological and Biochemical Characterization of Anthrax Lethal Factor, a Proteolytic Inhibitor of MEK Signaling Pathways. Methods Enzymol. 2008, 438, 355–365. [Google Scholar] [CrossRef]
- Ivarsson, M.E.; Leroux, J.C.; Castagner, B. Targeting bacterial toxins. Angew. Chem. Int. Ed. 2012, 51, 4024–4045. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Ventola, C. The antibiotic resistance crisis: Part 1: Causes and threats. Pharmacol. Ther. 2015, 40, 277–283. [Google Scholar]
- Yoshimatsu, K.; Koide, H.; Hoshino, Y.; Shea, K.J. Preparation of abiotic polymer nanoparticles for sequestration and neutralization of a target peptide toxin. Nat. Protoc. 2015, 10, 595–604. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Ruthel, G.; Stegmann, C.M.; Panchal, R.G.; Nguyen, T.L.; Hermone, A.R.; Stafford, R.G.; Lane, D.J.; Kenny, T.A.; McGrath, C.F.; et al. Inhibition of metalloprotease botulinum serotype A from a pseudo-peptide binding mode to a small molecule that is active in primary neurons. J. Biol. Chem. 2007, 282, 5004–5014. [Google Scholar] [CrossRef]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Shoop, W.L.; Xiong, Y.; Wiltsie, J.; Woods, A.; Guo, J.; Pivnichny, J.V.; Felcetto, T.; Michael, B.F.; Bansal, A.; Cummings, R.T.; et al. Anthrax lethal factor inhibition. Proc. Natl. Acad. Sci. USA 2005, 102, 7958–7963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pelish, T.M.; McClain, M.S. Dominant-negative inhibitors of the Clostridium perfringens ε-toxin. J. Biol. Chem. 2009, 284, 29446–29453. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Fang, R.H.; Copp, J.; Luk, B.T.; Zhang, L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol. 2013, 8, 336–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scobie, H.M.; Thomas, D.; Marlett, J.M.; Destito, G.; Wigelsworth, D.J.; Collier, R.J.; Young, J.A.T.; Manchester, M. A soluble receptor decoy protects rats against anthrax lethal toxin challenge. J. Infect. Dis. 2005, 192, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Rainey, G.J.A.; Young, J.A.T. Antitoxins: Novel strategies to target agents of bioterrorism. Nat. Rev. Microbiol. 2004, 2, 721–726. [Google Scholar] [CrossRef]
- Tombola, F.; Oregna, F.; Brutsche, S.; Szabò, I.; Del Giudice, G.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Inhibition of the vacuolating and anion channel activities of the VacA toxin of Helicobacter pylori. FEBS Lett. 1999, 460, 221–225. [Google Scholar] [CrossRef]
- Bachmeyer, C.; Benz, R.; Barth, H.; Aktories, K.; Gilbert, M.; Popoff, M.R. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes and Vero cells: Inhibition of channel function by chloroquine and related compounds in vitro and intoxification in vivo. FASEB J. 2001, 15, 1658–1660. [Google Scholar] [CrossRef]
- Bachmeyer, C.; Orlik, F.; Barth, H.; Aktories, K.; Benz, R. Mechanism of C2-toxin inhibition by fluphenazine and related compounds: Investigation of their binding kinetics to the C2II-channel using the current noise analysis. J. Mol. Biol. 2003, 333, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Kronhardt, A.; Beitzinger, C.; Barth, H.; Benz, R. Chloroquine Analog Interaction with C2- and Iota-Toxin in Vitro and in Living Cells. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.O.; Lea, E.J.; Finkelstein, A. Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions. Single-channel analysis. J. Gen. Physiol. 1990, 96, 921–942. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.O.; Koehler, T.M.; Collier, R.J.; Finkelstein, A. Anthrax toxin: Channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl. Acad. Sci. USA 1989, 86, 2209–2213. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Droogmans, G.; Nilius, B. Potent block of volume-activated chloride currents in endothelial cells by the uncharged form of quinine and quinidine. Br. J. Pharmacol. 1996, 118, 1869–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballestero, J.A. Effects of Quinine, Quinidine and Chloroquine on α9α10 Nicotinic Cholinergic Receptors. Mol. Pharmacol. 2005, 68, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.T.; Rhee, C.K.; Lee, C.S.; Park, Y.S.; Choi, D.C. Ototoxicity of salicylate, nonsteroidal antiinflammatory drugs, and quinine. Otolaryngol. Clin. North Am. 1993, 26, 791–810. [Google Scholar] [PubMed]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Cheng, Y.; Ma, X.; Wei, Y.; Wei, X.-W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef]
- Wilke, G.A.; Wardenburg, J.B. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus α-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Seilie, E.S.; Bubeck Wardenburg, J. Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity. Semin. Cell Dev. Biol. 2017, 72, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Schwiering, M.; Husmann, M.; Hellmann, N. P2X-receptor antagonists inhibit the interaction of S. aureus hemolysin A with membranes. Toxins 2017, 9. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, D.; Zhang, Y.; Dong, J.; Wang, J.; Niu, X. Molecular modeling reveals the novel inhibition mechanism and binding mode of three natural compounds to Staphylococcal α-hemolysin. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Lee, J.H.; Cho, M.H.; Lee, J. Red wines and flavonoids diminish Staphylococcus aureus virulence with anti-biofilm and anti-hemolytic activities. Biofouling 2014, 31, 1–11. [Google Scholar] [CrossRef]
- Qiu, J.; Niu, X.; Dong, J.; Wang, D.; Wang, J.; Li, H.; Luo, M.; Li, S.; Feng, H.; Deng, X. Baicalin protects mice from Staphylococcus aureus pneumonia via inhibition of the cytolytic activity of α-hemolysin. J. Infect. Dis. 2012, 206, 292–301. [Google Scholar] [CrossRef]
- Zhao, X.; Li, H.; Wang, J.; Guo, Y.; Liu, B.; Deng, X.; Niu, X. Verbascoside Alleviates Pneumococcal Pneumonia by Reducing Pneumolysin Oligomers. Mol. Pharmacol. 2016, 89, 376–387. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, B.; Liu, S.; Wang, L.; Wang, J. Anticytotoxin Effects of Amentoflavone to Pneumolysin. Biol. Pharm. Bull. 2017, 40, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Arzanlou, M.; Bohlooli, S. Inhibition of streptolysin O by allicin—An active component of garlic. J. Med. Microbiol. 2010, 59, 1044–1049. [Google Scholar] [CrossRef]
- Wang, J.; Qiu, J.; Tan, W.; Zhang, Y.; Wang, H.; Zhou, X.; Liu, S.; Feng, H.; Li, W.; Niu, X.; et al. Fisetin inhibits Listeria monocytogenes virulence by interfering with the oligomerization of Listeriolysin O. J. Infect. Dis. 2015, 211, 1376–1387. [Google Scholar] [CrossRef]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moayeri, M.; Wiggins, J.F.; Lindeman, R.E.; Leppla, S.H. Cisplatin inhibition of anthrax lethal toxin. Antimicrob. Agents Chemother. 2006, 50, 2658–2665. [Google Scholar] [CrossRef] [PubMed]
- Sarac, M.S.; Peinado, J.R.; Leppla, S.H.; Lindberg, I. Protection against Anthrax Toxemia by Hexa-D-Arginine In Vitro and In Vivo. Infect. Immun. 2004, 72, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Thomas, D.; Gillespie, E.J.; Damoiseaux, R.; Rogers, J.; Saxe, J.P.; Huang, J.; Manchester, M.; Bradley, K.A. Amiodarone and bepridil inhibit anthrax toxin entry into host cells. Antimicrob. Agents Chemother. 2007, 51, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Dell’Aica, I.; Donà, M.; Tonello, F.; Piris, A.; Mock, M.; Montecucco, C.; Garbisa, S. Potent inhibitors of anthrax lethal factor from green tea. EMBO Rep. 2004, 5, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numa, M.M.D.; Lee, L.V.; Hsu, C.C.; Bower, K.E.; Wong, C.H. Identification of novel anthrax lethal factor inhibitors generated by combinatorial Pictet-Spengler reaction followed by screening in situ. ChemBioChem 2005, 6, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Hermone, A.R.; Nguyen, T.L.; Wong, T.Y.; Schwarzenbacher, R.; Schmidt, J.; Lane, D.; McGrath, C.; Turk, B.E.; Burnett, J.; et al. Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 2004, 11, 67–72. [Google Scholar] [CrossRef]
- Tonello, F.; Seveso, M.; Marin, O.; Mock, M.; Montecucco, C. Screening inhibitors of anthrax lethal factor. Nature 2002, 418, 386. [Google Scholar] [CrossRef]
- Xiong, Y.; Wiltsie, J.; Woods, A.; Guo, J.; Pivnichny, J.V.; Tang, W.; Bansal, A.; Cummings, R.T.; Cunningham, B.R.; Friedlander, A.M.; et al. The discovery of a potent and selective lethal factor inhibitor for adjunct therapy of anthrax infection. Bioorg. Med. Chem. Lett. 2006, 16, 964–968. [Google Scholar] [CrossRef]
- Lena, G.; Trapani, J.A.; Sutton, V.R.; Ciccone, A.; Browne, K.A.; Smyth, M.J.; Denny, W.A.; Spicer, J.A. Dihydrofuro[3,4-c]pyridinones as inhibitors of the cytolytic effects of the pore-forming glycoprotein perforin. J. Med. Chem. 2008, 51, 7614–7624. [Google Scholar] [CrossRef]
- Lyons, D.M.; Huttunen, K.M.; Browne, K.A.; Ciccone, A.; Trapani, J.A.; Denny, W.A.; Spicer, J.A. Inhibition of the cellular function of perforin by 1-amino-2,4-dicyanopyrido[1,2-a]benzimidazoles. Bioorg. Med. Chem. 2011, 19, 4091–4100. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.A.; Huttunen, K.M.; Miller, C.K.; Denny, W.A.; Ciccone, A.; Browne, K.A.; Trapani, J.A. Inhibition of the pore-forming protein perforin by a series of aryl-substituted isobenzofuran-1(3H)-ones. Bioorg. Med. Chem. 2012, 20, 1319–1336. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.K.; Huttunen, K.M.; Denny, W.A.; Jaiswal, J.K.; Ciccone, A.; Browne, K.A.; Trapani, J.A.; Spicer, J.A. Diarylthiophenes as inhibitors of the pore-forming protein perforin. Bioorg. Med. Chem. Lett. 2016, 26, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Kodama, T.; Okahata, Y.; Shea, K.J. Peptide imprinted polymer nanoparticles: A plastic antibody. J. Am. Chem. Soc. 2008, 130, 15242–15243. [Google Scholar] [CrossRef] [PubMed]
- Mahon, C.S.; Fulton, D.A. Mimicking nature with synthetic macromolecules capable of recognition. Nat. Chem. 2014, 6, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Koide, H.; Furuya, K.; Haberaecker, W.W.; Lee, S.; Kodama, T.; Kanazawa, H.; Oku, N.; Shea, K.J. The rational design of a synthetic polymer nanoparticle that neutralizes a toxic peptide in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, K.; Yamazaki, T.; Hoshino, Y.; Rose, P.E.; Epstein, L.F.; Miranda, L.P.; Tagari, P.; Beierle, J.M.; Yonamine, Y.; Shea, K.J. Epitope discovery for a synthetic polymer nanoparticle: A new strategy for developing a peptide tag. J. Am. Chem. Soc. 2014, 136, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Germolec, D.R.; Weaver, J.L. Evaluation of nanoparticle immunotoxicity. Nat. Nanotechnol. 2009, 4, 411–414. [Google Scholar] [CrossRef]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggard, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle–protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Petros, R.A.; Desimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, J.A.; Maassen, C.B.M.; Leppla, S.H.; Brasky, K.; Patterson, J.L.; Iverson, B.L.; Georgiou, G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat. Biotechnol. 2002, 20, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Zettervall, O.; Sjöquist, J.; Waldenström, J.; Winblad, S. Serological activity in myeloma type globulins. Clin. Exp. Immunol. 1966, 1, 213–222. [Google Scholar] [PubMed]
- Seligmann, M.; Danon, F.; Basch, A.; Bernard, J. IgG myeloma cryoglobulin with antistreptolysin activity. Nature 1968, 220, 711–712. [Google Scholar] [CrossRef]
- Michaelsen, T.E.; Forre, O.; Hoyby, A.; Lea, T. Streptolysin O neutralizing capacity and idiotypic properties of fragments, subunits and reassociated H and L chains from three human IgG monoclonal proteins. Mol. Immunol. 1980, 17, 1143–1153. [Google Scholar] [CrossRef]
- Darji, A.; Niebuhr, K.; Hense, M.; Wehland, J.; Chakraborty, T. Neutralizing monoclonal antibodies against listeriolysin: Mapping of epitopes involved in pore formation. Infect. Immun. 1996, 64, 2356–2358. [Google Scholar]
- Nato, F.; Reich, K.; Lhopital, S.; Rouyre, S.; Geoffroy, C.; Mazie, J.C.; Cossart, P. Production and characterization of neutralizing and nonneutralizing monoclonal antibodies against listeriolysin O. Infect. Immun. 1991, 59, 4641–4646. [Google Scholar] [Green Version]
- Jacobs, T.; Cima-Cabal, M.D.; Darji, A.; Méndez, F.J.; Vázquez, F.; Jacobs, A.A.; Shimada, Y.; Ohno-Iwashita, Y.; Weiss, S.; de los Toyos, J.R. The conserved undecapeptide shared by thiol-activated cytolysins is involved in membrane binding. FEBS Lett. 1999, 459, 463–466. [Google Scholar] [CrossRef] [Green Version]
- Praper, T.; Beseničar Podlesnik, M.; Istinič, H.; Podlesek, Z.; Metkar, S.S.; Froelich, C.J.; Anderluh, G. Human perforin permeabilizing activity, but not binding to lipid membranes, is affected by pH. Mol. Immunol. 2010, 47, 2492–2504. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Moayeri, M.; Purcell, R. Monoclonal antibody therapies against anthrax. Toxins 2011, 3, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Little, S.F.; Leppla, S.H.; Cora, E. Production and Characterization of Monoclonal Antibodies to the Protective Antigen Component of Bacillus anthracis Toxin. Infect. Immun. 1988, 56, 1807–1813. [Google Scholar] [PubMed]
- Little, S.F.; Novak, J.M.; Lowe, J.R.; Leppla, S.H.; Singh, Y.; Klimpel, K.R.; Lidgerding, B.C.; Friedlanderl, A.M. Characterization of lethal factor binding and cell receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology 1996, 142, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Migone, T.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.; Clagett, M.; Li, J.; Jones, S.; Pincus, S.; D’Alia, G.; Nardone, L.; Babin, M.; Spitalny, G.; Casey, L. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect. Immun. 2005, 73, 795–802. [Google Scholar] [CrossRef]
- Brossier, F.; Le, M.; Landier, A.; Lafaye, P. Functional Analysis of Bacillus anthracis Protective Antigen by Using Neutralizing Monoclonal Antibodies. Infect. Immun. 2004, 72, 6313–6317. [Google Scholar] [CrossRef]
- Wang, F.; Ruther, P.; Jiang, I.; Sawada-Hirai, R.; Sun, S.M.; Nedellec, R.; Morrow, P.R.; Kang, A.S. Human monoclonal antibodies that neutralize anthrax toxin by inhibiting heptamer assembly. Hum. Antib. 2004, 13, 105–110. [Google Scholar] [CrossRef]
- Vitale, L.; Blanset, D.; Lowy, I.; O’Neill, T.; Goldstein, J.; Little, S.F.; Andrews, G.P.; Dorough, G.; Taylor, R.K.; Keler, T. Prophylaxis and therapy of inhalational anthrax by a novel monoclonal antibody to protective antigen that mimics vaccine-induced immunity. Infect. Immun. 2006, 74, 5840–5847. [Google Scholar] [CrossRef]
- Peterson, J.W.; Comer, J.E.; Noffsinger, D.M.; Wenglikowski, A.; Walberg, K.G.; Chatuev, B.M.; Chopra, A.K.; Stanberry, L.R.; Kang, A.S.; Scholz, W.W.; et al. Human monoclonal anti-protective antigen antibody completely protects rabbits and is synergistic with ciprofloxacin in protecting mice and guinea pigs against inhalation anthrax. Infect. Immun. 2006, 74, 1016–1024. [Google Scholar] [CrossRef]
- Karginov, V.A.; Robinson, T.M.; Riemenschneider, J.; Golding, B.; Kennedy, M.; Shiloach, J.; Alibek, K. Treatment of anthrax infection with combination of ciprofloxacin and antibodies to protective antigen of Bacillus anthracis. FEMS Immunol. Med. Microbiol. 2004, 40, 71–74. [Google Scholar] [CrossRef]
- Orth, P.; Xiao, L.; Hernandez, L.D.; Reichert, P.; Sheth, P.R.; Beaumont, M.; Yang, X.; Murgolo, N.; Ermakov, G.; Dinunzio, E.; et al. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J. Biol. Chem. 2014, 289, 18008–18021. [Google Scholar] [CrossRef] [PubMed]
- Harshman, S.; Alouf, J.E.; Siffert, O.; Baleux, F. Reaction of staphylococcal alpha-toxin with peptide-induced antibodies. Infect. Immun. 1989, 57, 3856–3862. [Google Scholar] [PubMed]
- Ragle, B.E.; Wardenburg, J.B. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 2009, 77, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.S.; Cover, T.L. Functional analysis of neutralizing antibodies against Clostridium perfringens epsilon-toxin. Infect. Immun. 2007, 75, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Percival, D.A.; Shuttleworth, A.D.; Williamson, E.D.; Kelly, D.C. Anti-idiotypic antibody-induced protection against Clostridium perfringens type D. Infect. Immun. 1990, 58, 2487–2492. [Google Scholar] [PubMed]
- Rouha, H.; Badarau, A.; Visram, Z.C.; Battles, M.B.; Prinz, B.; Magyarics, Z.; Nagy, G.; Mirkina, I.; Stulik, L.; Zerbs, M.; et al. Five birds, one stone: Neutralization of α-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. mAbs 2015, 7, 243–254. [Google Scholar] [CrossRef]
- Schlesinger, B.C.; Cheng, L. Characterization of a novel monoclonal antibody against human perforin using transfected cell lines. Immunology 1994, 81, 291–295. [Google Scholar]
- Liu, J.K.H. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. 2014, 3, 113–116. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy: Protein engineering strategies to improve exposure in solid tumors. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Hoogenboomt, H.R.; Griffithst, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrebaeck, C.A.K. Antibodies in diagnostics - from immunoassays to protein chips. Immunol. Today 2000, 21, 379–382. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Plückthun, A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005, 23, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Skerra, A. Alternative non-antibody scaffolds for molecular recognition. Curr. Opin. Biotechnol. 2007, 18, 295–304. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef]
- Gebauer, M.; Skerra, A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 2009, 13, 245–255. [Google Scholar] [CrossRef]
- Liu, J.L.; Anderson, G.P.; Goldman, E.R. Isolation of anti-toxin single domain antibodies from a semi-synthetic spiny dogfish shark display library. BMC Biotechnol. 2007, 7. [Google Scholar] [CrossRef]
- He, M.; Taussig, M.J. Emerging Technologies for Antibody Selection. In Handbook of Therapeutic Antibodies; Dübel, S., Reichert, J.M., Eds.; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2014; pp. 393–405. [Google Scholar]
- Wild, M.A.; Xin, H.; Maruyama, T.; Nolan, M.J.; Calveley, P.M.; Malone, J.D.; Wallace, M.R.; Bowdish, K.S. Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat. Biotechnol. 2003, 21, 1305–1306. [Google Scholar] [CrossRef]
- Ding, G.; Chen, X.; Zhu, J.; Duesbery, N.S.; Cheng, X.; Cao, B. A human/murine chimeric fab antibody neutralizes anthrax lethal toxin in vitro. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Zheng, F.; Xiong, S.; Hu, D.; Lv, H.; Tang, Q.; Yang, J.; Feng, Z.; Wang, C.; Zhu, J. Preparation and evaluation of human-murine chimeric antibody against protective antigen of Bacillus anthracis. Int. J. Mol. Sci. 2014, 15, 18496–18507. [Google Scholar] [CrossRef] [PubMed]
- Pelat, T.; Hust, M.; Laffly, E.; Condemine, F.; Bottex, C.; Vidal, D.; Lefranc, M.P.; Dübel, S.; Thullier, P. High-affinity, human antibody-like antibody fragment (single-chain variable fragment) neutralizing the lethal factor (LF) of Bacillus anthracis by inhibiting protective antigen-LF complex formation. Antimicrob. Agents Chemother. 2007, 51, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Gómez, I.; Miranda-Ríos, J.; Arenas, I.; Grande, R.; Becerril, B.; Bravo, A. Identification of scFv Molecules that Recognize Loop 3 of Domain II and Domain III of Cry1Ab Toxin from Bacillus thuringiensis. In Proceedings of the 6th Pacific Rim Conference on the Biotechnology of Bacillus thuringiensis and its Environmental Impact, Victoria, BC, Canada, 30 October–3 November 2005; pp. 12–14. [Google Scholar]
- Goldman, E.R.; Andersen, G.P.; Liu, J.L.; Delehanty, J.B.; Sherwood, L.J.; Osborn, L.E.; Cummins, L.B.; Hayhurst, A. Facile generation of heat-stable antiviral and antitoxin single domain antibodies from a semisynthetic llama library. Anal. Chem. 2006, 78, 8245–8255. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Anderson, G.P.; Delehanty, J.B.; Baumann, R.; Hayhurst, A.; Goldman, E.R. Selection of cholera toxin specific IgNAR single-domain antibodies from a naive shark library. Mol. Immunol. 2007, 44, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Hussack, G.; Arbabi-Ghahroudi, M.; Van Faassen, H.; Songer, J.G.; Ng, K.K.S.; MacKenzie, R.; Tanha, J. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J. Biol. Chem. 2011, 286, 8961–8976. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.; Eichhoff, A.M.; Schumacher, L.; Strysio, M.; Menzel, S.; Schwan, C.; Alzogaray, V.; Zylberman, V.; Seman, M.; Brandner, J.; et al. Selection of Nanobodies that Block the Enzymatic and Cytotoxic Activities of the Binary Clostridium Difficile Toxin CDT. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Yang, N.J.; Liu, D.V.; Sklaviadis, D.; Gui, D.Y.; Vander Heiden, M.G.; Wittrup, K.D. Antibody-Mediated Neutralization of Perfringolysin O for Intracellular Protein Delivery. Mol. Pharm. 2015, 12, 1992–2000. [Google Scholar] [CrossRef] [Green Version]
- Mammen, M.; Choi, S.; Whitesides, G.M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Crini, G. Review: A history of cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- Karginov, V.A.; Nestorovich, E.M.; Schmidtmann, F.; Robinson, T.M.; Yohannes, A.; Fahmi, N.E.; Bezrukov, S.M.; Hecht, S.M. Inhibition of S. aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg. Med. Chem. 2007, 15, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Ragle, B.E.; Karginov, V.A.; Wardenburg, J.B. Prevention and treatment of Staphylococcus aureus pneumonia with a β-cyclodextrin derivative. Antimicrob. Agents Chemother. 2010, 54, 298–304. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.C.; Caballero, A.R.; Balzli, C.L.; Tang, A.; O’Callaghan, R.J. Chemical inhibition of alpha-toxin, a key corneal virulence factor of Staphylococcus aureus. Invest. Ophthalmol. Vis. Sci. 2009, 50, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- Backer, M.V.; Patel, V.; Jehning, B.T.; Claffey, K.P.; Karginov, V.A.; Backer, J.M. Inhibition of anthrax protective antigen outside and inside the cell. Antimicrob. Agents Chemother. 2007, 51, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Yohannes, A.; Robinson, T.M.; Fahmi, N.E.; Schmidtmann, F.; Hecht, S.M.; Bezrukov, S.M. Search for cyclodextrin-based inhibitors of anthrax toxins: Synthesis, structural features, and relative activities. Antimicrob. Agents Chemother. 2006, 50, 3740–3753. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Yohannes, A.; Robinson, T.M.; Fahmi, N.E.; Alibek, K.; Hecht, S.M. β-Cyclodextrin derivatives that inhibit anthrax lethal toxin. Bioorg. Med. Chem. 2006, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Robinson, T.M.; Leppla, S.H.; Karginov, V.A. In vivo efficacy of beta-cyclodextrin derivatives against anthrax lethal toxin. Antimicrob. Agents Chemother. 2008, 52, 2239–2241. [Google Scholar] [CrossRef]
- Bezrukov, S.M.; Liu, X.; Karginov, V.A.; Wein, A.N.; Leppla, S.H.; Popoff, M.R.; Barth, H.; Nestorovich, E.M. Interactions of high-affinity cationic blockers with the translocation pores of B. anthracis, C. botulinum, and C. perfringens binary toxins. Biophys. J. 2012, 103, 1208–1217. [Google Scholar] [CrossRef]
- Roeder, M.; Nestorovich, E.M.; Karginov, V.A.; Schwan, C.; Aktories, K.; Barth, H. Tailored Cyclodextrin Pore Blocker Protects Mammalian Cells from Clostridium difficile Binary Toxin CDT. Toxins 2014, 6, 2097–2114. [Google Scholar] [CrossRef]
- Nestorovich, E.M.; Karginov, V.A.; Popoff, M.R.; Bezrukov, S.M.; Barth, H. Tailored ß-cyclodextrin blocks the translocation pores of binary exotoxins from C. botulinum and C. perfringens and protects cells from intoxication. PLoS ONE 2011, 6, e23927. [Google Scholar] [CrossRef]
- Mourez, M.; Kane, R.S.; Mogridge, J.; Metallo, S.; Deschatelets, P.; Sellman, B.R.; Whitesides, G.M.; Collier, R.J. Designing a polyvalent inhibitor of anthrax toxin. Nat. Biotechnol. 2001, 19, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Padala, C.; Poon, V.; Saraph, A.; Basha, S.; Kate, S.; Tao, K.; Mogridge, J.; Kane, R.S. Statistical pattern matching facilitates the design of polyvalent inhibitors of anthrax and cholera toxins. Nat. Biotechnol. 2006, 24, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.; Smith Korsholm, K.; Andersen, P.; Agger, E.M. Cationic liposomes as vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 513–521. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, M.L.; Rabasco, A.M. Charged liposomes as carriers to enhance the permeation through the skin. Expert Opin. Drug Deliv. 2011, 8, 857–871. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Smith Korsholm, K.; Andersen, P.; Perrie, Y.; Christensen, D. Liposomal vaccine delivery systems. Expert Opin. Drug Deliv. 2011, 8, 505–519. [Google Scholar] [CrossRef]
- Förstner, P.; Bayer, F.; Kalu, N.; Felsen, S.; Förtsch, C.; Aloufi, A.; Ng, D.Y.W.; Weil, T.; Nestorovich, E.M.; Barth, H. Cationic PAMAM dendrimers as pore-blocking binary toxin inhibitors. Biomacromolecules 2014, 15, 2461–2474. [Google Scholar] [CrossRef]
- Yamini, G.; Kalu, N.; Nestorovich, E.M. Impact of dendrimer terminal group chemistry on blockage of the anthrax toxin channel: A single molecule study. Toxins 2016, 8. [Google Scholar] [CrossRef]
- Kitov, P.I.; Sadowska, J.M.; Mulvey, G.; Armstrong, G.D.; Ling, H.; Pannu, N.S.; Read, R.J.; Bundle, D.R. Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 2000, 403, 669–672. [Google Scholar] [CrossRef]
- Basha, S.; Rai, P.; Poon, V.; Saraph, A.; Gujraty, K.; Go, M.Y.; Sadacharan, S.; Frost, M.; Mogridge, J.; Kane, R.S. Polyvalent inhibitors of anthrax toxin that target host receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 13509–13513. [Google Scholar] [CrossRef] [Green Version]
- Yamini, G.; Nestorovich, E.M. Multivalent Inhibitors of Channel-Forming Bacterial Toxins. In Current topics in microbiology and immunology; Barth, H., Ed.; Springer International Publishing: Basel, Switzerland, 2016. [Google Scholar]
- Rummel, A.; Karnath, T.; Henke, T.; Bigalke, H.; Binz, T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 2004, 279, 30865–30870. [Google Scholar] [CrossRef]
- Dong, M.; Richards, D.A.; Goodnough, M.C.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R. Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J. Cell Biol. 2003, 162, 1293–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.M.; Wang, J.-L.; Kang, L.; Gao, S.; Liu, Y.; Hu, T.M. Construction and analysis of high-complexity ribosome display random peptide libraries. PLoS ONE 2008, 3, e2092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, W.; Chen, Y.; Escajadillo, T.; Ungerleider, J.; Fang, R.H.; Christman, K.; Nizet, V.; Zhang, L. Self-Assembled Colloidal Gel Using Cell Membrane-Coated Nanosponges as Building Blocks. ACS Nano 2017, 11, 11923–11930. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.D.; Neill, D.R.; Becker, K.A.; Gore, S.; Bricio-Moreno, L.; Ziobro, R.; Edwards, M.J.; Mühlemann, K.; Steinmann, J.; Kleuser, B.; et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 2015, 33, 81–88. [Google Scholar] [CrossRef]
- Sharma, S.; Thomas, D.; Marlett, J.; Manchester, M.; Young, J.A.T. Efficient neutralization of antibody-resistant forms of anthrax toxin by a soluble receptor decoy inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1210–1212. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Luk, B.T.; Hu, C.J.; Zhang, L. Engineered nanoparticles mimicking cell membranes for toxin neutralization. Adv. Drug Deliv. Rev. 2015, 90, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Gao, W.; Thamphiwatana, S.; Luk, B.T.; Angsantikul, P.; Zhang, Q.; Hu, C.J.; Fang, R.H.; Copp, J.A.; Pornpattananangkul, D.; et al. Hydrogel Retaining Toxin-Absorbing Nanosponges for Local Treatment of Methicillin-Resistant Staphylococcus aureus Infection. Adv. Mater. 2015, 27, 3437–3443. [Google Scholar] [CrossRef]
- Cai, C.; Che, J.; Xu, L.; Guo, Q.; Kong, Y.; Fu, L.; Xu, J.; Cheng, Y.; Chen, W. Tumor endothelium marker-8 based decoys exhibit superiority over capillary morphogenesis protein-2 based decoys as anthrax toxin inhibitors. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Wycoff, K.L.; Belle, A.; Deppe, D.; Schaefer, L.; MacLean, J.M.; Haase, S.; Trilling, A.K.; Liu, S.; Leppla, S.H.; Geren, I.N.; et al. Recombinant anthrax toxin receptor-Fc fusion proteins produced in plants protect rabbits against inhalational anthrax. Antimicrob. Agents Chemother. 2011, 55, 132–139. [Google Scholar] [CrossRef]
- Narat, M.; Maček, P.; Kotnik, V.; Sedmak, B. The humoral and cellular immune response to a lipid attenuated pore-forming toxin from the sea anemone Actinia equina L. Toxicon 1994, 32, 65–71. [Google Scholar] [CrossRef]
- Turk, T.; Maček, P.; Gubenšek, F. Chemical modification of equinatoxin II, a lethal and cytolytic toxin from the sea anemone Actinia equina L. Toxicon 1989, 27, 375–384. [Google Scholar] [CrossRef]
- Polyzos, A.; Alderton, M.R.; Dawson, R.M.; Hartley, P.G. Biofunctionalized surfactant mesophases as polyvalent inhibitors of cholera toxin. Bioconjug. Chem. 2007, 18, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Vinion-Dubiel, A.D.; McClain, M.S.; Czajkowsky, D.M.; Iwamoto, H.; Ye, D.; Cao, P.; Schraw, W.; Szabo, G.; Blanke, S.R.; Shao, Z.; et al. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 1999, 274, 37736–37742. [Google Scholar] [CrossRef] [PubMed]
- Sellman, B.R.; Mourez, M.; Collier, R.J. Dominant-negative mutants of a toxin subunit: An approach to therapy of anthrax. Science 2001, 292, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Khanna, H.; Chopra, A.P.; Mehra, V. A Dominant Negative Mutant of Bacillus anthracis Protective Antigen Inhibits Anthrax Toxin Action in Vivo. J. Biol. Chem. 2001, 276, 22090–22094. [Google Scholar] [CrossRef] [PubMed]
- Mourez, M.; Yan, M.; Lacy, D.B.; Dillon, L.; Bentsen, L.; Marpoe, A.; Maurin, C.; Hotze, E.; Wigelsworth, D.; Pimental, R.; et al. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 13803–13808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Y.; Roehrl, M.H. Anthrax vaccine design: Strategies to achieve comprehensive protection against spore, bacillus, and toxin. Med. Immunol. 2005, 4. [Google Scholar] [CrossRef]
- Cao, S.; Guo, A.; Liu, Z.; Tan, Y.; Wu, G.; Zhang, C.; Zhao, Y.; Chen, H. Investigation of new dominant-negative inhibitors of anthrax protective antigen mutants for use in therapy and vaccination. Infect. Immun. 2009, 77, 4679–4687. [Google Scholar] [CrossRef]
- Yan, M.; Collier, R.J. Characterization of Dominant-Negative Forms of Anthrax Protective Antigen. Mol. Med. 2003, 9, 46–51. [Google Scholar] [CrossRef]
- Rodríguez-Almazán, C.; Zavala, L.E.; Muñoz-Garay, C.; Jiménez-Juárez, N.; Pacheco, S.; Masson, L.; Soberón, M.; Bravo, A. Dominant negative mutants of Bacillus thuringiensis Cry1Ab toxin function as anti-toxins: Demonstration of the role of oligomerization in toxicity. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Wai, S.N.; Westermark, M.; Oscarsson, J.; Jass, J.; Maier, E.; Benz, R.; Uhlin, B.E. Characterization of dominantly negative mutant ClyA cytotoxin proteins in Escherichia coli. J. Bacteriol. 2003, 185, 5491–5499. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. A dominant-negative therapy for anthrax. Nat. Med. 2001, 7, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Olsen, K.J.; Cheng, L.; Fox, W.M.; Hruban, R.H.; Podackt, E.R. Immunohistochemical Identification of Cytotoxic Lymphocytes Using Human Perforin Monoclonal Antibody. Am. J. Pathol. 1992, 140, 1025–1030. [Google Scholar] [PubMed]
- Portman, J.L.; Huang, Q.; Reniere, M.L.; Iavarone, A.T.; Portnoy, D.A. Activity of the pore-forming virulence factor Listeriolysin O is reversibly inhibited by naturally occurring S-glutathionylation. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Geisberg, M.; Trapani, J.A.; Dupont, B. Monoclonal antibodies detecting discrete epitopes of human perforin. Tissue Antigens 1990, 35, 229–233. [Google Scholar] [CrossRef] [PubMed]
- McComb, R.C.; Martchenko, M. Neutralizing antibody and functional mapping of Bacillus anthracis protective antigen—The first step toward a rationally designed anthrax vaccine. Vaccine 2016, 34, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Hugo, F.; Reichwein, J.; Arvand, M.; Krämer, S.; Bhakdi, S. Use of a monoclonal antibody to determine the mode of transmembrane pore formation by streptolysin O. Infect. Immun. 1986, 54, 641–645. [Google Scholar] [PubMed]
- Majd, S.; Yusko, E.C.; Billeh, Y.N.; Macrae, M.X.; Yang, J.; Mayer, M. Applications of biological pores in nanomedicine, sensing, and nanoelectronics. Curr. Opin. Biotechnol. 2010, 21, 439–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, N.; Osaki, T.; Takeuchi, S. Membrane protein-based biosensors. J. R. Soc. Interface 2018, 15, 20170952. [Google Scholar] [CrossRef]
- Robertson, J.W.F.; Rodrigues, C.G.; Stanford, V.M.; Rubinson, K.A.; Krasilnikov, O.V.; Kasianowicz, J.J. Single-molecule mass spectrometry in solution using a solitary nanopore. Proc. Natl. Acad. Sci. USA 2007, 104, 8207–8211. [Google Scholar] [CrossRef] [Green Version]
- Rauf, S.; Zhang, L.; Ali, A.; Liu, Y.; Li, J. Label-Free Nanopore Biosensor for Rapid and Highly Sensitive Cocaine Detection in Complex Biological Fluids. ACS Sensors 2017, 2, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Montana, V.; Grubišić, V.; Stout, R.F.; Parpura, V.; Gu, L.Q. Nanopore sensing of botulinum toxin type B by discriminating an enzymatically cleaved peptide from a synaptic protein synaptobrevin 2 derivative. ACS Appl. Mater. Interfaces 2015, 7, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Edwards, S.; Schmidt, J. Research highlights: Nanopore protein detection and analysis. Lab. Chip. 2015, 15, 3424–3427. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Wu, H.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ayub, M.; Bayley, H. Engineered transmembrane pores. Curr. Opin. Chem. Biol. 2016, 34, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayley, H. Nanopore sequencing: From imagination to reality. Clin. Chem. 2015, 61, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Misra, N.; Martinez, J.A.; Huang, S.J.; Wang, Y.; Stroeve, P.; Grigoropoulos, C.P. Bioelectronic silicon nanowire devices using functional membrane proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 13780–13784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provoda, C.J.; Stier, E.M.; Lee, K.D. Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 2003, 278, 35102–35108. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omersa, N.; Podobnik, M.; Anderluh, G. Inhibition of Pore-Forming Proteins. Toxins 2019, 11, 545. https://doi.org/10.3390/toxins11090545
Omersa N, Podobnik M, Anderluh G. Inhibition of Pore-Forming Proteins. Toxins. 2019; 11(9):545. https://doi.org/10.3390/toxins11090545
Chicago/Turabian StyleOmersa, Neža, Marjetka Podobnik, and Gregor Anderluh. 2019. "Inhibition of Pore-Forming Proteins" Toxins 11, no. 9: 545. https://doi.org/10.3390/toxins11090545
APA StyleOmersa, N., Podobnik, M., & Anderluh, G. (2019). Inhibition of Pore-Forming Proteins. Toxins, 11(9), 545. https://doi.org/10.3390/toxins11090545