Activation of the IRE1α Arm, but not the PERK Arm, of the Unfolded Protein Response Contributes to Fumonisin B1-Induced Hepatotoxicity


Abstract

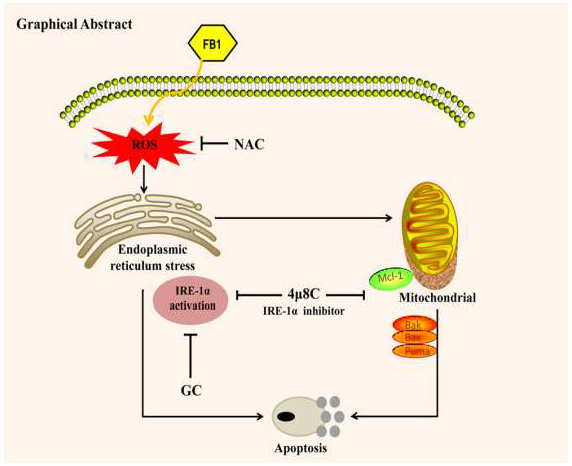{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
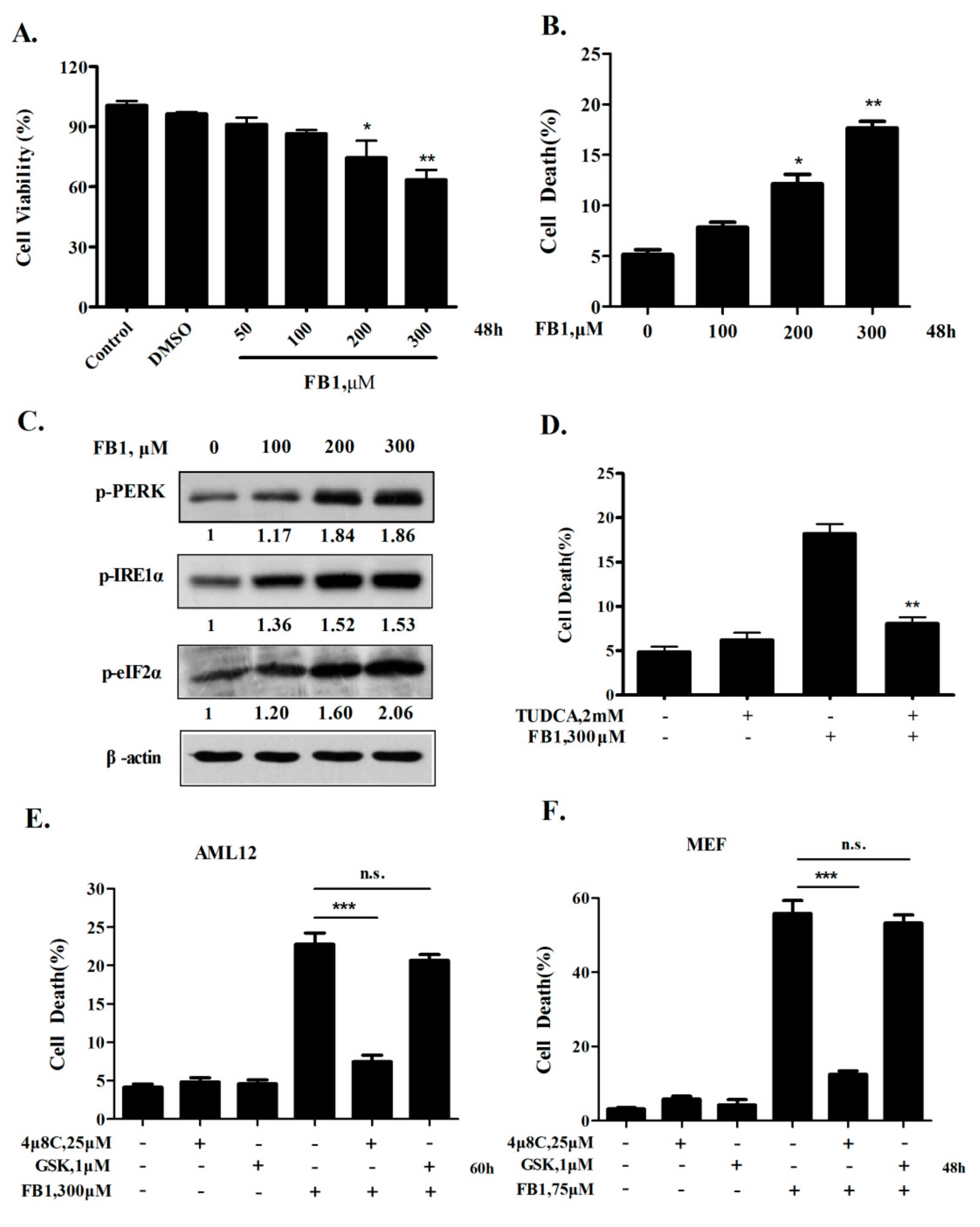
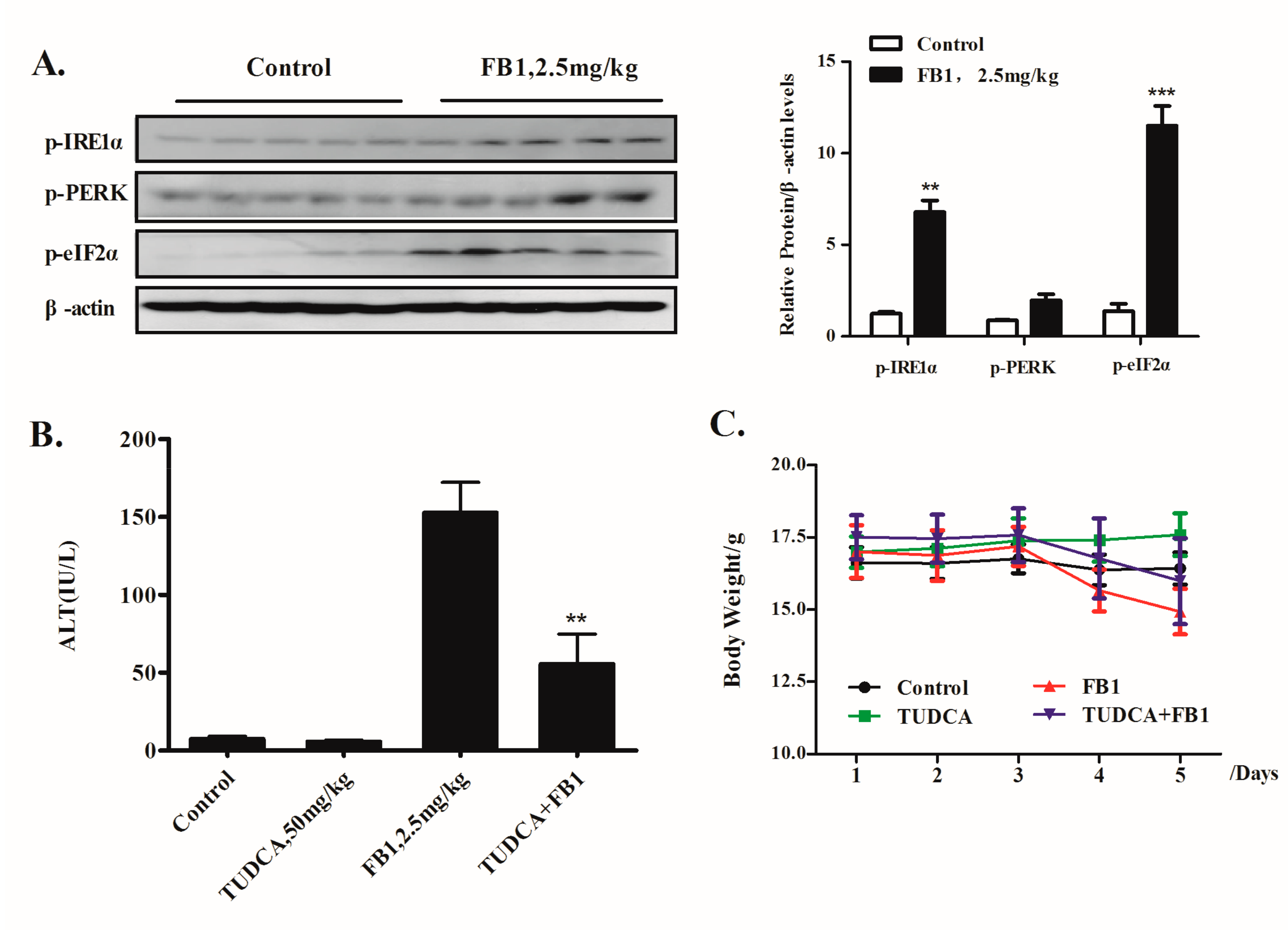
2.1. Activation of IRE1α but not the PERK Axis of ER Stress Response Contributes to FB1-Induced ER Stress-Mediated Hepatocyte Toxicity
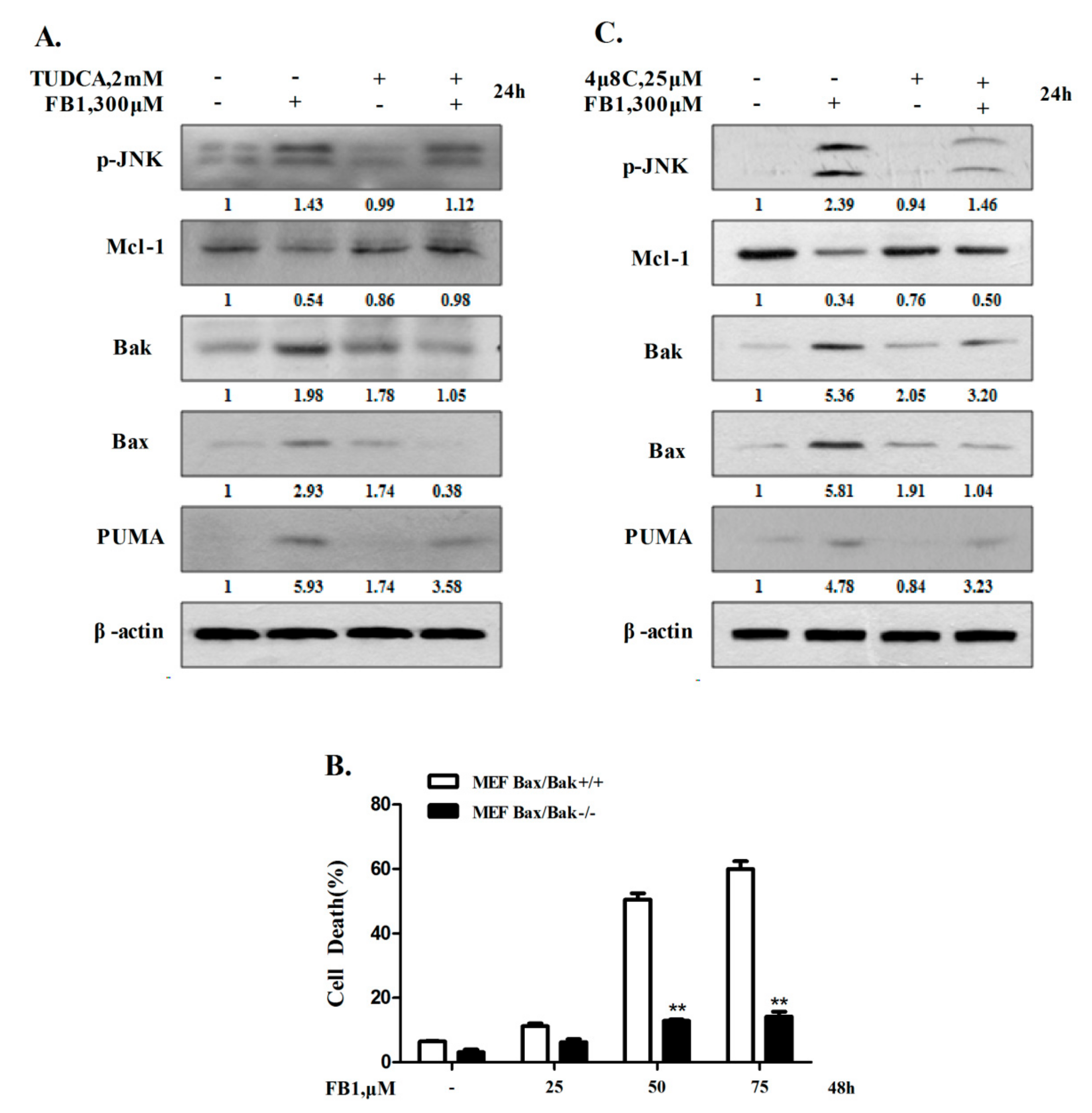
2.2. IRE1α-Mediated Activation of Mitochondrial Pathway Plays an Important Role in Apoptosis Induction by FB1 in Liver Cells
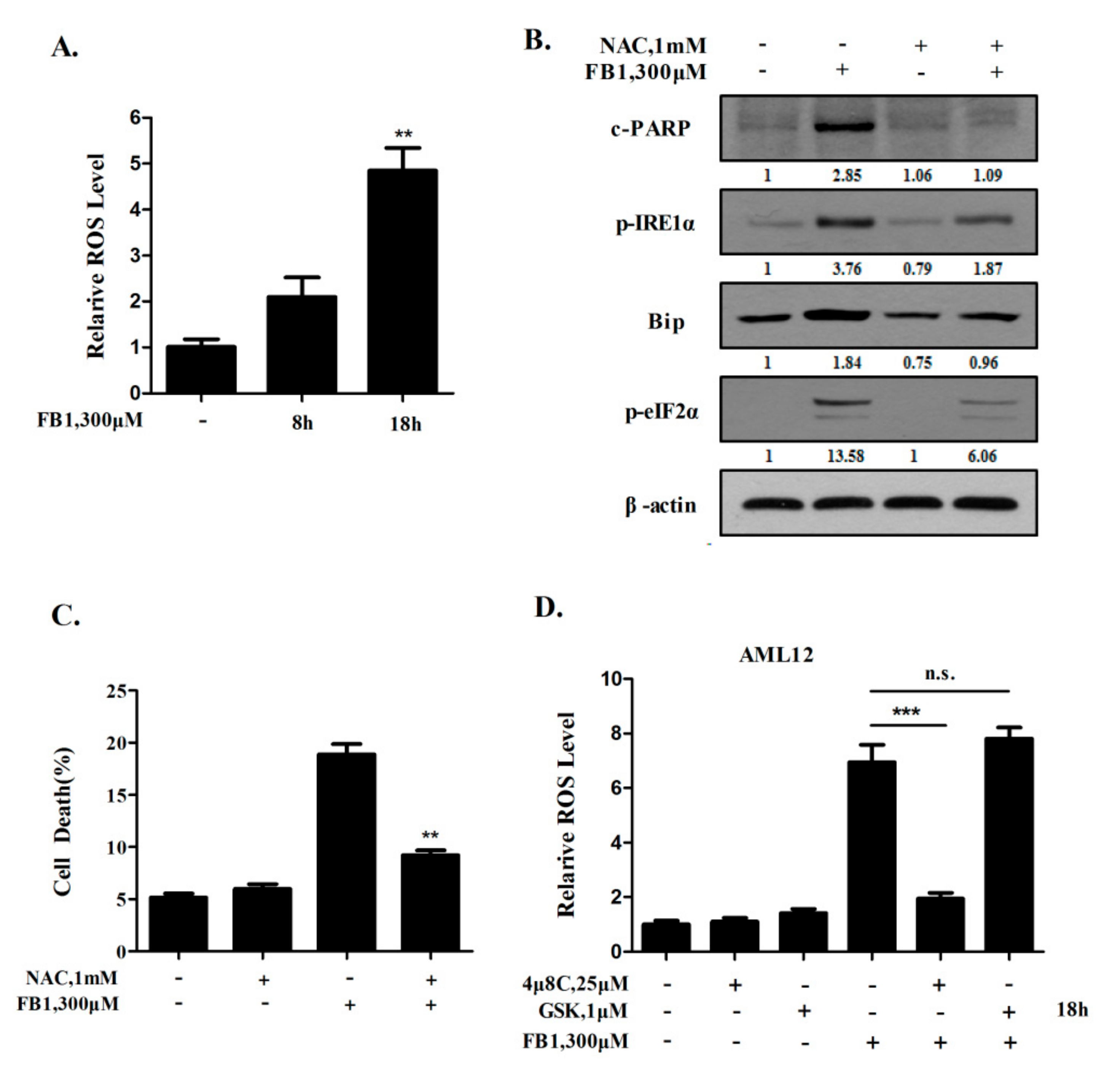
2.3. A Positive Feedback Loop Exists between ER Stress Activation and ROS Generation Induced by FB1
2.4. Inhibition of ER Stress by TUDCA Leads to a Significant Reduction of FB1-Induced Liver Injury In Vivo
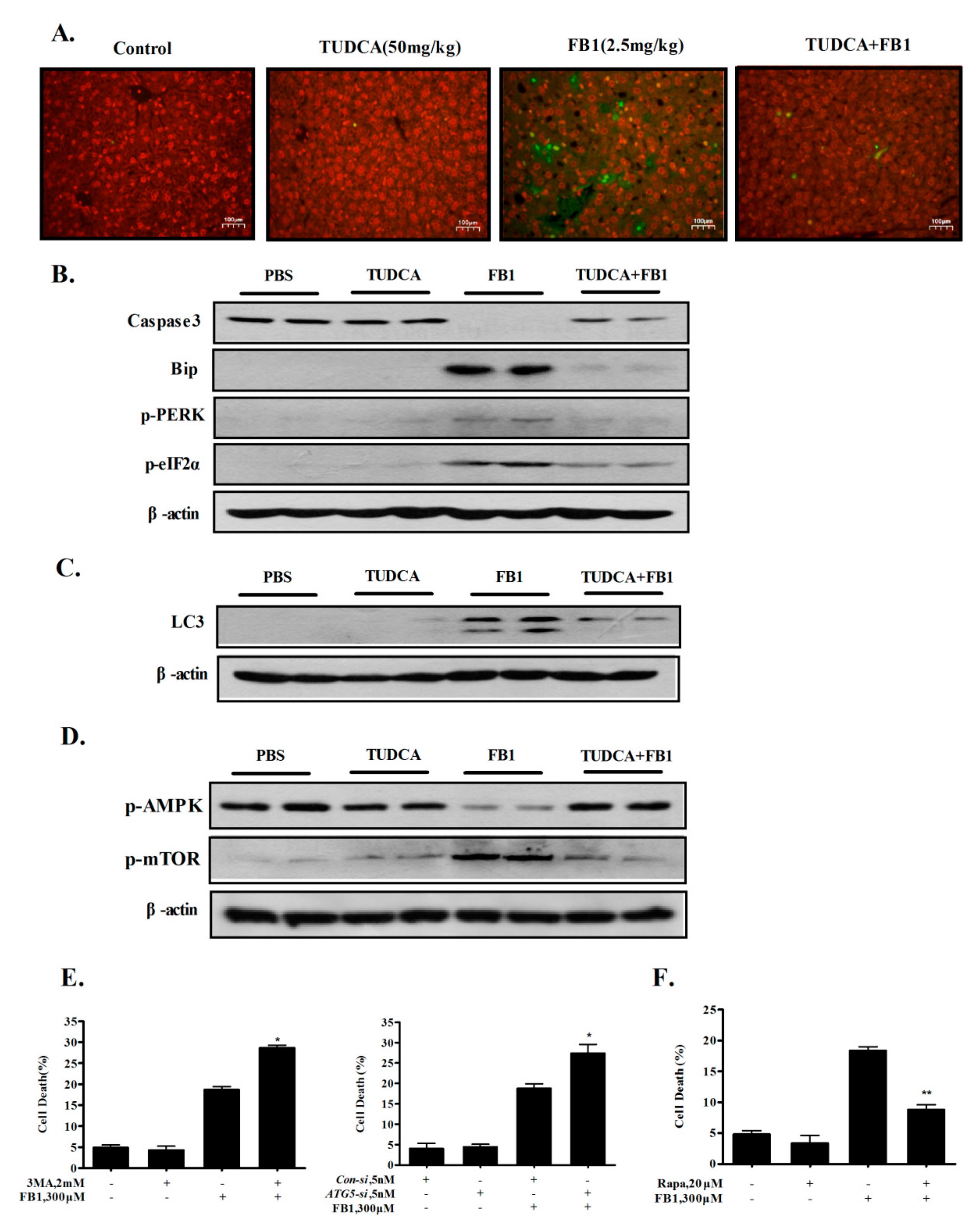
2.5. Inhibition of ER Stress by TUDCA Inhibits FB1-Induced Hepatic Apoptosis and Autophagy In Vivo
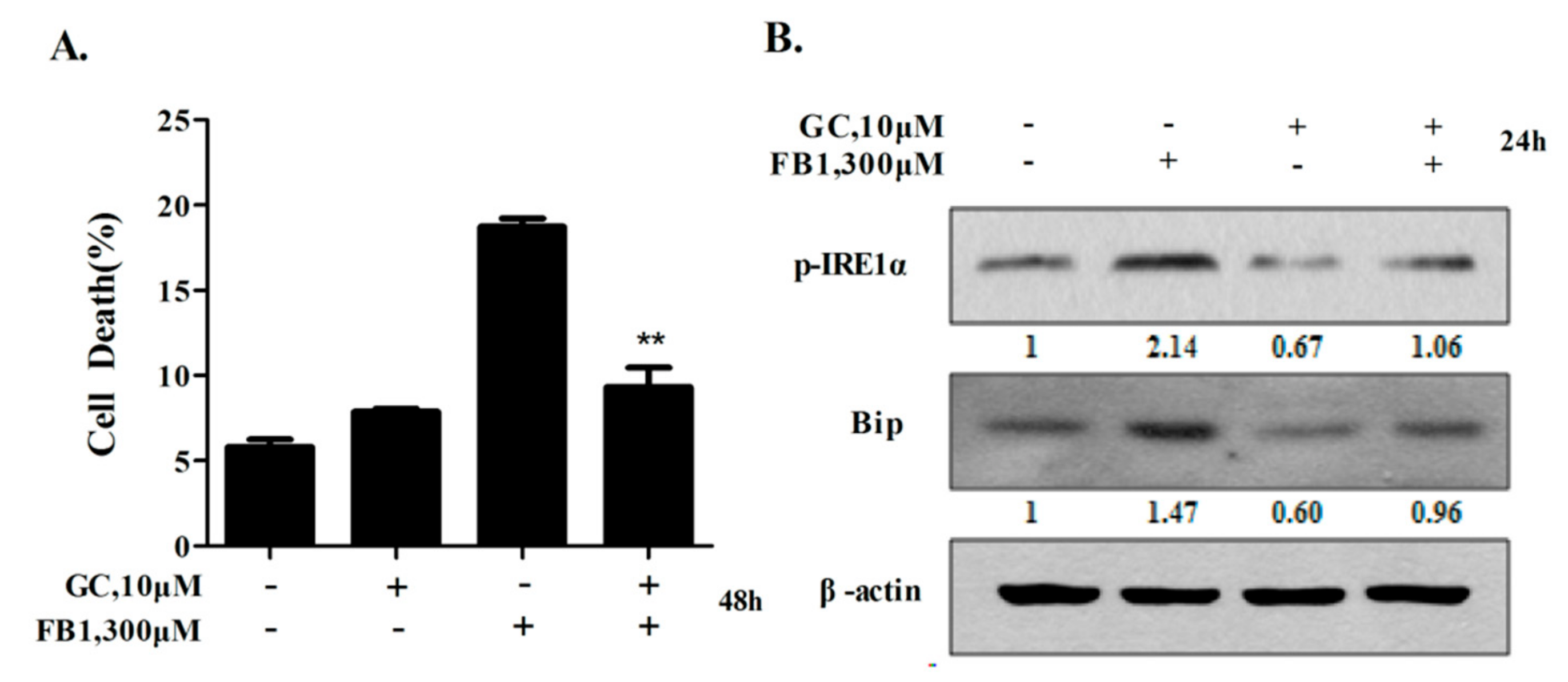
2.6. Glycyrol Prevents FB1-Induced Apoptosis in AML12 Cells Through Inactivating IRE1α
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Cell Culture and Treatments
5.3. Apoptosis Evaluation
5.4. Proliferation Assay
5.5. Western Blotting
5.6. Assessment of Reactive Oxygen Species
5.7. RNA Interference
5.8. Animals and Treatments
5.9. ALT Measurement
5.10. Histochemical and Immunohistochemical Staining
5.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Marciniak, S.J. Endoplasmic reticulum stress in lung disease. Eur. Respir. Rev. 2017, 26, 170018. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yoo, W.H.; Chae, H.J. ER Stress and Autophagy. Curr. Mol. Med. 2015, 15, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Aspects Med. 2019, 66, 49–61. [Google Scholar] [CrossRef]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar]
- Song, S.; Tan, J.; Miao, Y.; Zhang, Q. Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 2018, 233, 3867–3874. [Google Scholar] [CrossRef]
- Dara, L.; Ji, C.; Kaplowitz, N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011, 53, 1752–1763. [Google Scholar] [CrossRef]
- Moretti, A.; Logrieco, A.F.; Susca, A. Mycotoxins: An Underhand Food Problem. In Mycotoxigenic Fungi: Methods and Protocols; Moretti, A., Susca, A., Eds.; Springer: New York, NY, USA, 2017; pp. 3–12. ISBN 978-1-4939-6707-0. [Google Scholar]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef]
- Gu, Q.; Tahir, A.H.; Zhang, H.; Huang, H.; Ji, T.; Sun, X.; Wu, L.; Wu, H.; Gao, X. Involvement of FvSet1 in Fumonisin B1 Biosynthesis, Vegetative Growth, Fungal Virulence, and Environmental Stress Responses in Fusarium verticillioides. Toxins 2017, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Thiel, P.G.; Marasas, W.F.; Sydenham, E.W.; Shephard, G.S.; Gelderblom, W.C.; Nieuwenhuis, J.J. Survey of fumonisin production by Fusarium species. Appl. Environ. Microbiol. 1991, 57, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Guo, X.; Li, J.; Fan, L.; Hu, H. Fumonisin B1 induces autophagic cell death via activation of ERN1-MAPK8/9/10 pathway in monkey kidney MARC-145 cells. Arch. Toxicol. 2016, 90, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P.; Kang, S.C. Endoplasmic reticulum stress-mediated autophagy activation attenuates fumonisin B1 induced hepatotoxicity in vitro and in vivo. Food Chem. Toxicol. 2017, 110, 371–382. [Google Scholar] [CrossRef]
- Sato, H.; Shiba, Y.; Tsuchiya, Y.; Saito, M.; Kohno, K. 4μ8C Inhibits Insulin Secretion Independent of IRE1α RNase Activity. Cell Struct. Funct. 2017, 70, 61–70. [Google Scholar] [CrossRef]
- Liu, M.; Shi, G.; Zhou, A.; Rupert, C.E.; Coulombe, K.L.K.; Dudley, S.C. Activation of the unfolded protein response downregulates cardiac ion channels in human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 117, 62–71. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Stockmann-Juvala, H.; Mikkola, J.; Naarala, J.; Loikkanen, J.; Elovaara, E.; Savolainen, K. Oxidative Stress Induced by Fumonisin B1 in Continuous Human and Rodent Neural Cell Cultures. Free Radic. Res. 2004, 38, 933–942. [Google Scholar] [CrossRef]
- Abbès, S.; Ben Salah-Abbès, J.; Jebali, R.; Younes, R.B.; Oueslati, R. Interaction of aflatoxin B1 and fumonisin B1 in mice causes immunotoxicity and oxidative stress: Possible protective role using lactic acid bacteria. J. Immunotoxicol. 2016, 13, 46–54. [Google Scholar] [CrossRef]
- Hojjati, M.R.; Li, Z.; Zhou, H.; Tang, S.; Huan, C.; Ooi, E.; Lu, S.; Jiang, X. Effect of Myriocin on Plasma Sphingolipid Metabolism and Atherosclerosis in apoE-deficient Mice *. J. Biol. Chem. 2005, 280, 10284–10289. [Google Scholar] [CrossRef]
- Boppana, N.B.; Kraveka, J.M.; Rahmaniyan, M.; Li, L.I.; Bielawska, A.; Bielawski, J.; Pierce, J.S.; Delor, J.S.; Zhang, K.; Korbelik, M.; et al. Fumonisin B1 Inhibits Endoplasmic Reticulum Stress Associated-apoptosis After FoscanPDT Combined with C6-Pyridinium Ceramide or Fenretinide. Anticancer Res. 2017, 37, 455–463. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, S.H.; Singh, M.P.; Sharma, C.; Kang, S.C. Fumonisin B1 actuates oxidative stress-associated colonic damage via apoptosis and autophagy activation in murine model. J. Biochem. Mol. Toxicol. 2018, 32, e22161. [Google Scholar] [CrossRef] [PubMed]
- Chuturgoon, A.A.; Phulukdaree, A.; Moodley, D. Fumonisin B 1 inhibits apoptosis in HepG2 cells by inducing Birc-8/ILP-2. Toxicol. Lett. 2015, 235, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-J.R.; Kim, H.; Oh, S.; Lee, J.-G.; Kee, M.; Ko, H.-J.; Kweon, M.-N.; Won, K.-J.; Baek, S.H. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ye, M. Chemical analysis of the Chinese herbal medicine Gan-Cao (licorice). J. Chromatogr. A 2009, 1216, 1954–1969. [Google Scholar] [CrossRef]
- Asl, M.N.; Hosseinzadeh, H. Review of Pharmacological Effects of Glycyrrhiza sp. and its Bioactive Compounds. Phyther. Res. 2008, 22, 709–724. [Google Scholar] [CrossRef]
- Qiao, X.; Liu, C.F.; Ji, S.; Lin, X.H.; Guo, D.A.; Ye, M. Simultaneous determination of five minor coumarins and flavonoids in Glycyrrhiza uralensis by solid-phase extraction and high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Planta Med. 2014, 80, 237–242. [Google Scholar] [CrossRef]
- Zhang, E.; Yin, S.; Song, X.; Fan, L.; Hu, H. Glycycoumarin inhibits hepatocyte lipoapoptosis through activation of autophagy and inhibition of ER stress/GSK-3-mediated mitochondrial pathway. Sci. Rep. 2016, 6, 38138. [Google Scholar] [CrossRef]
- Bro, L.E.; Kruyt, F.A.E.; Giaccone, G. Cell Death Independent of Caspases: A Review. Clin. Cancer Res. 2005, 11, 3155–3163. [Google Scholar]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging Mechanisms in Initiating and Terminating Autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Diao, X.; Li, N.; Liu, C. FB1-induced programmed cell death in hemocytes of Ostrinia furnacalis. Toxicon 2018, 146, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wang, Z.; Ganther, H.; Lü, J. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001, 61, 3062–3070. [Google Scholar] [PubMed]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Molecular and Cellular Endocrinology Metformin and resveratrol inhibit Drp1-mediated mitochondrial fi ssion and prevent ER stress-associated NLRP3 in fl ammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Moreira, S.; Fonseca, I.; João, M.; Rosa, A.; Lemos, L.; Rodrigues, E.; Neves, A.; Outeiro, T.F.; Maria, C.; Rodrigues, P.; et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 2017, 295, 77–87. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Zhang, E.; Yin, S.; Zhao, C.; Fan, L.; Hu, H. Activation of the IRE1α Arm, but not the PERK Arm, of the Unfolded Protein Response Contributes to Fumonisin B1-Induced Hepatotoxicity. Toxins 2020, 12, 55. https://doi.org/10.3390/toxins12010055
Liu X, Zhang E, Yin S, Zhao C, Fan L, Hu H. Activation of the IRE1α Arm, but not the PERK Arm, of the Unfolded Protein Response Contributes to Fumonisin B1-Induced Hepatotoxicity. Toxins. 2020; 12(1):55. https://doi.org/10.3390/toxins12010055
Chicago/Turabian StyleLiu, Xiaoyi, Enxiang Zhang, Shutao Yin, Chong Zhao, Lihong Fan, and Hongbo Hu. 2020. "Activation of the IRE1α Arm, but not the PERK Arm, of the Unfolded Protein Response Contributes to Fumonisin B1-Induced Hepatotoxicity" Toxins 12, no. 1: 55. https://doi.org/10.3390/toxins12010055
APA StyleLiu, X., Zhang, E., Yin, S., Zhao, C., Fan, L., & Hu, H. (2020). Activation of the IRE1α Arm, but not the PERK Arm, of the Unfolded Protein Response Contributes to Fumonisin B1-Induced Hepatotoxicity. Toxins, 12(1), 55. https://doi.org/10.3390/toxins12010055