Evaluation of Matrix Issues in the Applicability of the Neuro-2a Cell Based Assay on the Detection of CTX in Fish Samples


Abstract
1. Introduction
2. Results and Discussion
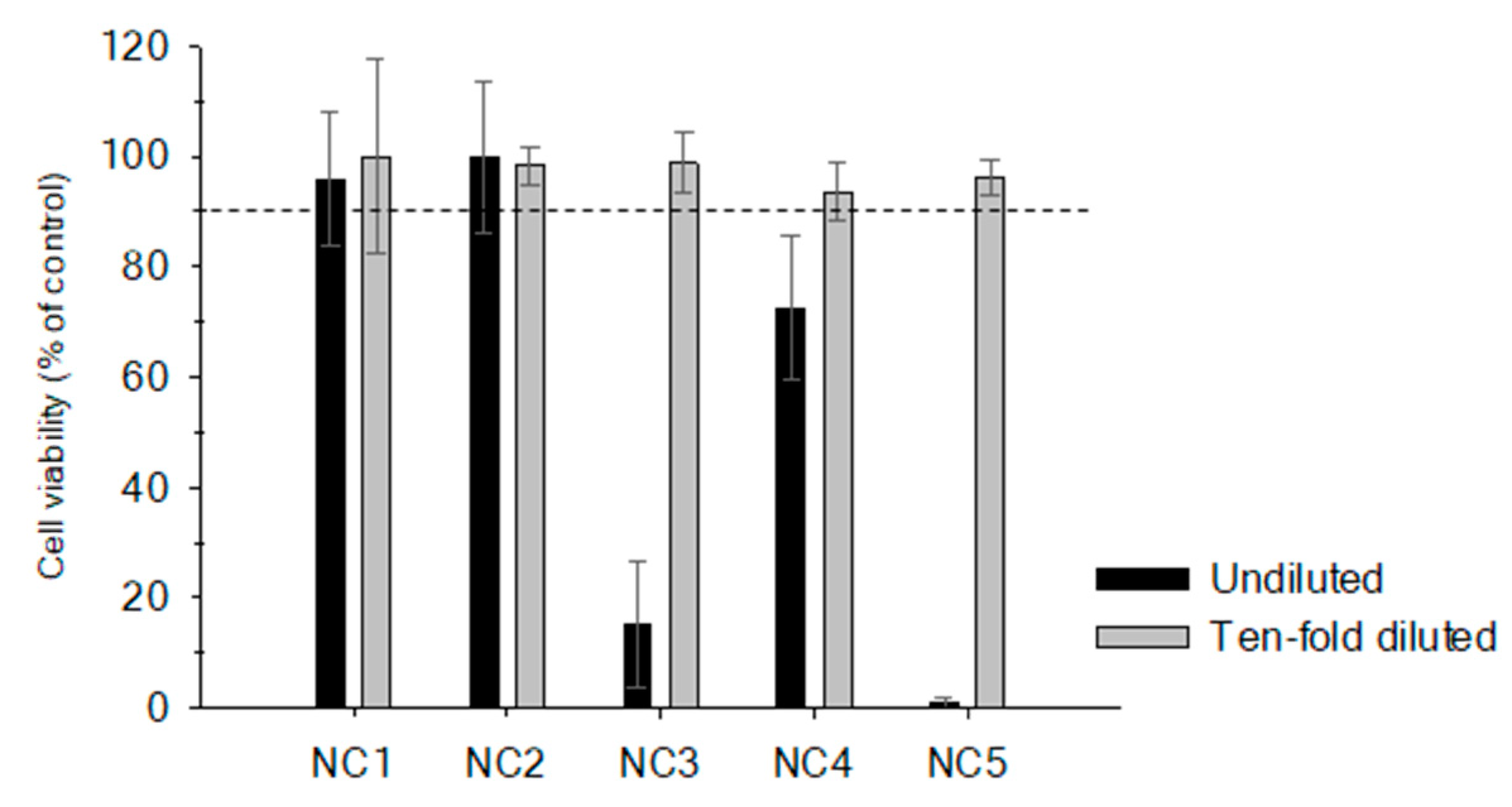
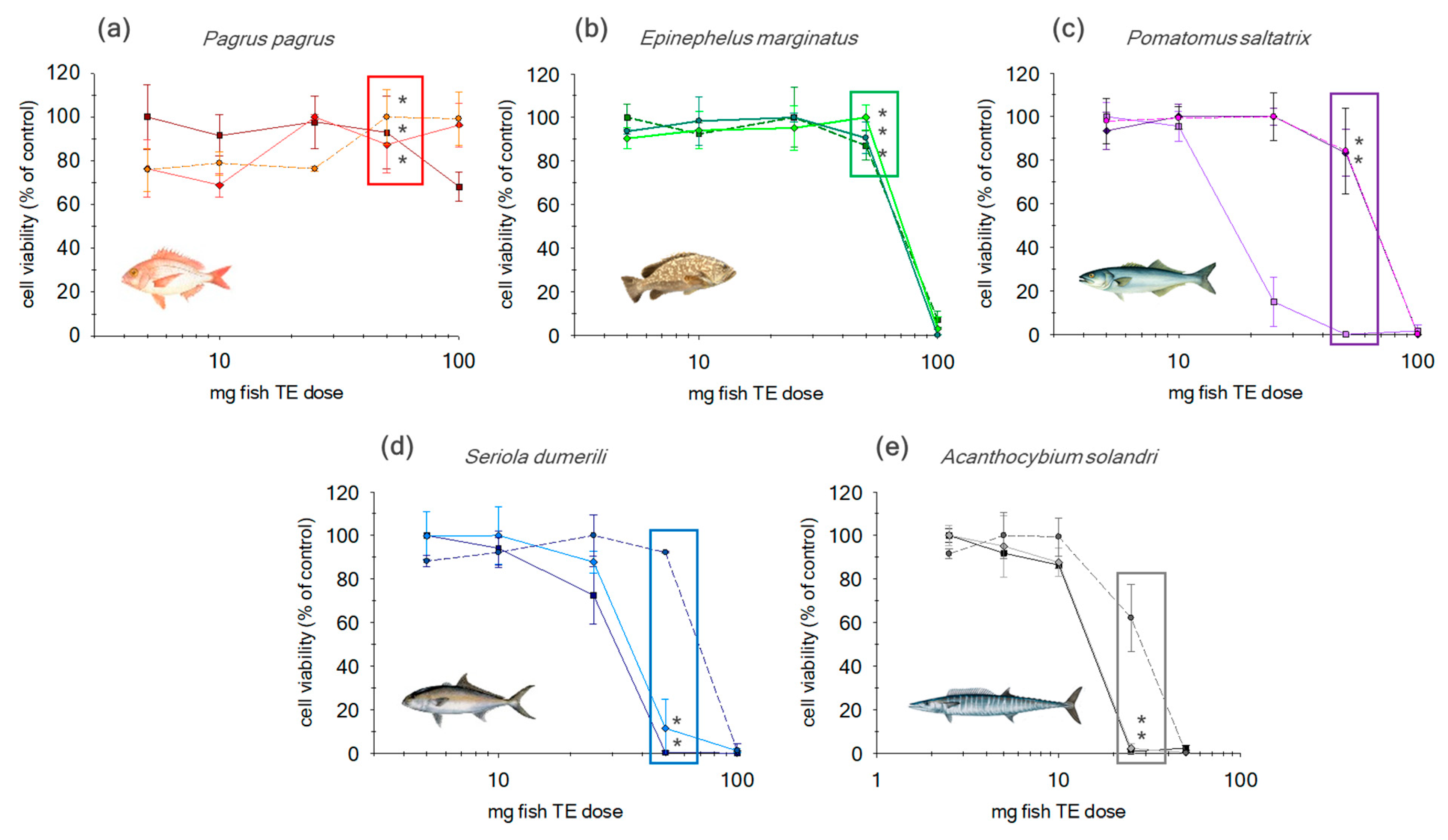
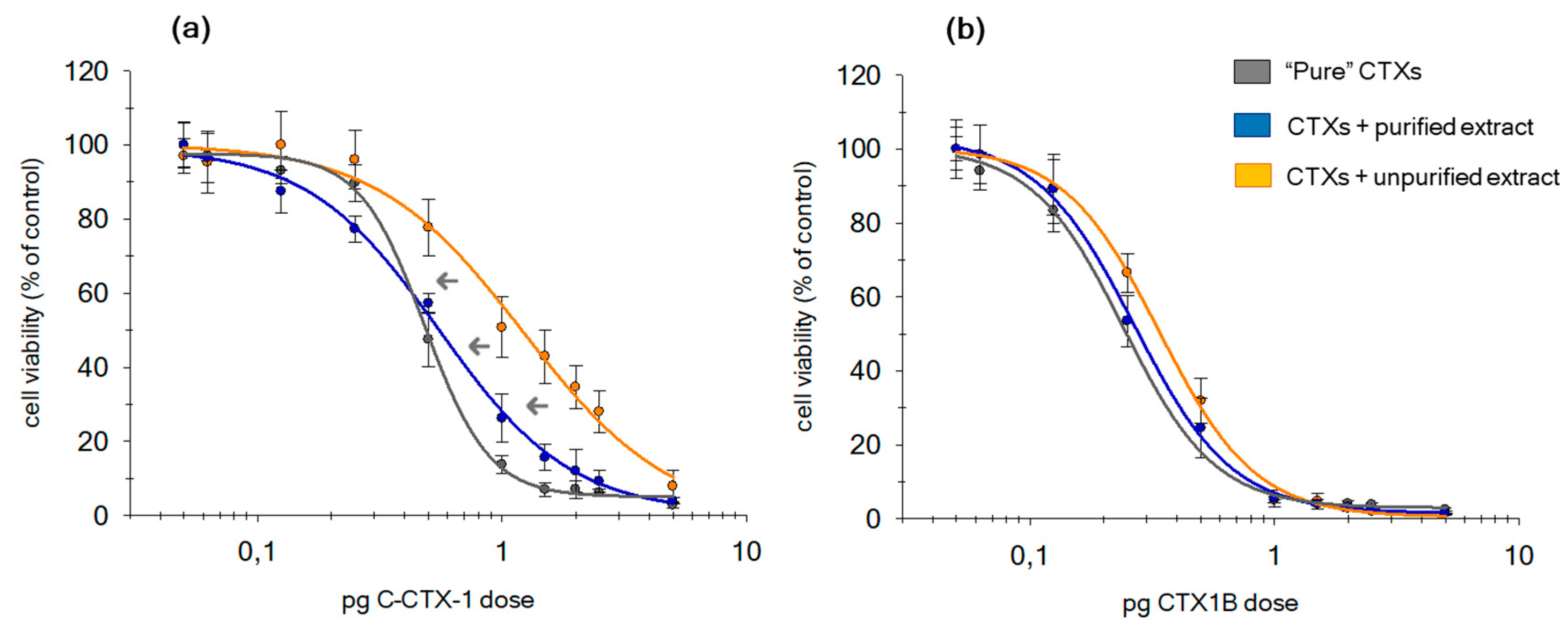
2.1. Matrix Effect in Non-Purified Sample Extracts
2.2. Evaluation of Additional Cleanup Steps
2.2.1. SPE-Florisil
2.2.2. SPE-Florisil + C18
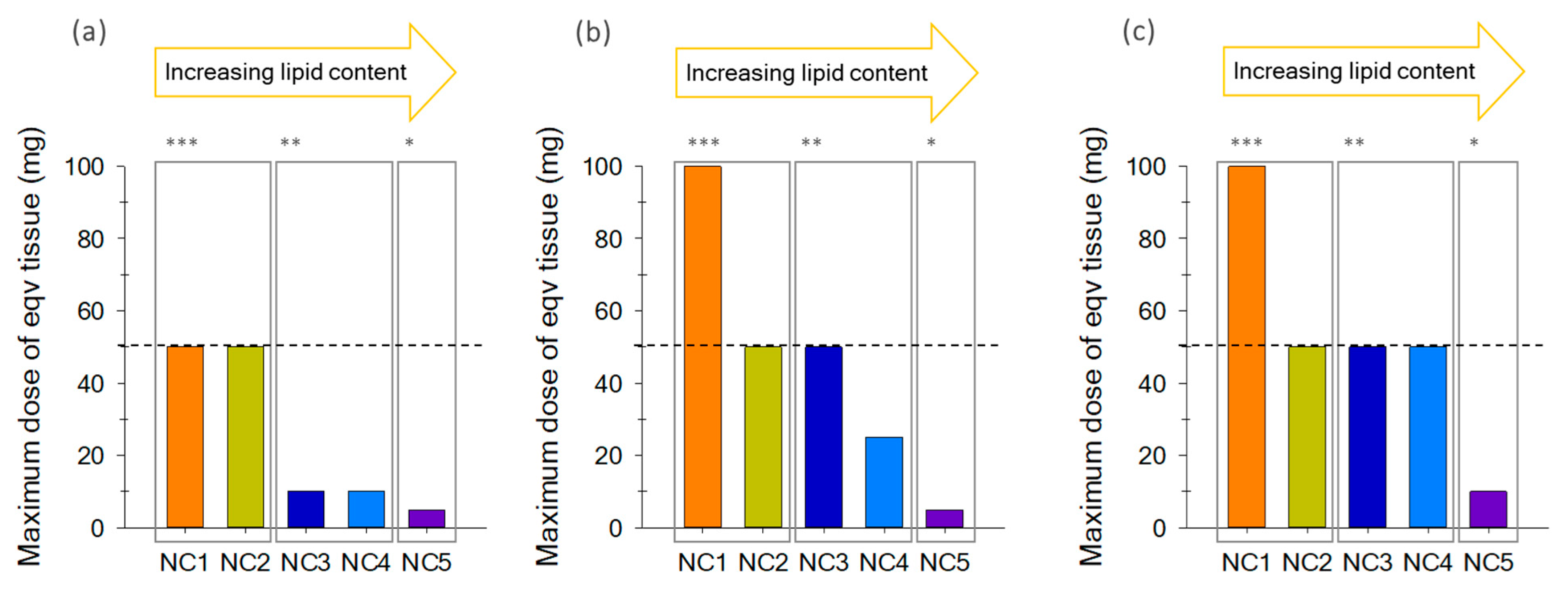
2.3. Optimum Maximum Tissue Dose Equivalent (MTDE)
2.4. Evaluation of the Toxicity of the Major CTX Analogues
2.4.1. CTX1B, CTX3C and C-CTX1 Toxic Potency in Neat Solution
2.4.2. CTX1B and C-CTX1 Effectiveness in the Presence of the Matrix
3. Conclusions
4. Materials and Methods
4.1. Standards and Reagents
4.2. Sample Pretreatment for the N2a-MTT Assay (and HPLC–MS/MS)
4.3. In Vitro N2a-MTT Assay
4.3.1. Maintenance of Culture
4.3.2. Cytotoxicity Cell Based assay (N2a-MTT Assay)
4.3.3. Measurement of Mitochondrial Activity
4.3.4. Analysis and statistical treatment of the data
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedman, M.A.; Fleming, L.E.; Fernandez, M.; Bienfang, P.; Schrank, K.; Dickey, R.; Bottein, M.Y.; Backer, L.; Ayyar, R.; Weisman, R.; et al. Ciguatera fish poisoning: Treatment, prevention and management. Mar. Drugs 2008, 6, 456–479. [Google Scholar] [CrossRef]
- Berdalet, E.; E Fleming, L.; Gowen, R.; Davidson, K.; Hess, P.; Backer, L.C.; Moore, S.K.; Hoagland, P.; Enevoldsen, H. Marine harmful algal blooms, human health and wellbeing: Challenges and opportunities in the 21st century. J. Mar. Boil. Assoc. UK 2015, 96, 61–91. [Google Scholar] [CrossRef]
- Lehane, L.; Lewis, R.J. Ciguatera: Recent advances but the risk remains. Int. J. Food Microbiol. 2000, 61, 91–125. [Google Scholar] [CrossRef]
- Fraga, S.; Rodríguez, F. Genus Gambierdiscus in the Canary Islands (NE Atlantic Ocean) with Description of Gambierdiscus silvae sp. nov., a New Potentially Toxic Epiphytic Benthic Dinoflagellate. Protist 2014, 165, 839–853. [Google Scholar] [CrossRef]
- Nishimura, T.; Sato, S.; Tawong, W.; Sakanari, H.; Uehara, K.; Shah, M.R.; Suda, S.; Yasumoto, T.; Taira, Y.; Yamaguchi, H.; et al. Genetic Diversity and Distribution of the Ciguatera-Causing Dinoflagellate Gambierdiscus spp. (Dinophyceae) in Coastal Areas of Japan. PLoS ONE 2013, 8, e60882. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Nakajima, I.; Bagnis, R.; Adachi, R. Finding of a dinoflagellate as a likely culprit of ciguatera. Bull. Jap. Soc. Sci. Fish 1977, 43, 1021–1026. [Google Scholar] [CrossRef]
- Gómez, F.; Qiu, D.; Lopes, R.; Lin, S. Fukuyoa paulensis gen. et sp. nov., a New Genus for the Globular Species of the Dinoflagellate Gambierdiscus (Dinophyceae). PLoS ONE 2015, 10, e0119676. [Google Scholar] [CrossRef] [PubMed]
- Bienfang, P.; Trapido-Rosenthal, H.; Laws, E.A. Bioaccumulation/Biomagnifications in Food Chains. Environ. Toxicol. 2012, 35–69. [Google Scholar] [CrossRef]
- Holmes, M.J.; Lewis, R.J. Toxin-producing dinoflagellates. In Perspectives in Molecular Toxicology; Menez, A., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2002; pp. 39–65. [Google Scholar]
- Litaker, R.W.; Vandersea, M.; Faust, M.A.; Kibler, S.; Nau, A.W.; Holland, W.C.; Chinain, M.; Holmes, M.J.; Tester, P.A. Global distribution of ciguatera causing dinoflagellates in the genus Gambierdiscus. Toxicon 2010, 56, 711–730. [Google Scholar] [CrossRef]
- Vernoux, J.-P.; Lewis, R.J. Isolation and characterisation of Caribbean ciguatoxins from the horse-eye jack (Caranx latus). Toxicon 1997, 35, 889–900. [Google Scholar] [CrossRef]
- Otero, P.; Pérez, S.; Alfonso, A.; Vale, C.; Rodrıíguez, P.; Gouveia, N.N.; Gouveia, N.; Delgado, J.; Vale, P.; Hirama, M. First toxin profile of ciguateric fish in Madeira Archipelago (Europe). Analyt. Biochem. 2010, 82, 6032–6039. [Google Scholar] [CrossRef] [PubMed]
- Arellano, J.L.P.; Luzardo, O.P.; Brito, A.P.; Cabrera, M.H.; Zumbado, M.; Carranza, C.; Angel-Moreno, A.; Dickey, R.W.; Boada, L.D. Ciguatera Fish Poisoning, Canary Islands. Emerg. Infect. Dis. 2005, 11, 1981–1982. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Vernoux, J.-P.; Brereton, I. Structure of Caribbean Ciguatoxin Isolated fromCaranx latus. J. Am. Chem. Soc. 1998, 120, 5914–5920. [Google Scholar] [CrossRef]
- Dickey, R.W.; Plakas, S.M. Ciguatera: A public health perspective. Toxicon 2010, 56, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Chain, E.P.O.C.I.T.F. Scientific Opinion on marine biotoxins in shellfish - Emerging toxins: Ciguatoxin group. EFSA J. 2010, 8, 1627. [Google Scholar] [CrossRef]
- Lombet, A.; Bidard, J.-N.; Lazdunski, M. Ciguatoxin and brevetoxins share a common receptor site on the neuronal voltage-dependent Na+ channel. FEBS Lett. 1987, 219, 355–359. [Google Scholar] [CrossRef]
- Dechraoui, M.-Y.; Naar, J.; Pauillac, S.; Legrand, A.-M. Ciguatoxins and brevetoxins, neurotoxic polyether compounds active on sodium channels. Toxicon 1999, 37, 125–143. [Google Scholar] [CrossRef]
- Van Dolah, F.M.; Finley, E.L.; Haynes, B.L.; Doucette, G.J.; Moeller, P.D.; Ramsdell, J.S. Development of rapid and sensitive high throughput pharmacologic assays for marine phycotoxins. Nat. Toxins 1994, 2, 189–196. [Google Scholar] [CrossRef]
- Manger, R.; Leja, L.; Lee, S.; Hungerford, J.; Wekell, M. Tetrazolium-Based Cell Bioassay for Neurotoxins Active on Voltage-Sensitive Sodium Channels: Semiautomated Assay for Saxitoxins, Brevetoxins, and Ciguatoxins. Anal. Biochem. 1993, 214, 190–194. [Google Scholar] [CrossRef]
- Manger, R.L.; Leja, L.S.; Lee, S.Y.; Hungerford, J.M.; Hokama, Y.; Dickey, R.W.; Granade, H.R.; Lewis, R.; Yasumoto, T.; Wekell, M.M. Detection of Sodium Channel Toxins: Directed Cytotoxicity Assays of Purified Ciguatoxins, Brevetoxins, Saxitoxins, and Seafood Extracts. J. AOAC Int. 1995, 78, 521–527. [Google Scholar] [CrossRef]
- Fairey, E.R.; Bottein Dechraoui, M.Y.; Sheets, M.F.; Ramsdell, J.S. Modification of the cell based assay for brevetoxinsusing human cardiac voltage dependent sodium channels expressed in HEK-293 cells. Biosens. Bioelectron. 2001, 16, 579–586. [Google Scholar] [CrossRef]
- Dechraoui, M.-Y.B.; Ramsdell, J.S. Type B brevetoxins show tissue selectivity for voltage-gated sodium channels: Comparison of brain, skeletal muscle and cardiac sodium channels. Toxicon 2003, 41, 919–927. [Google Scholar] [CrossRef]
- Dechraoui, M.-Y.B.; Wang, Z.; Turquet, J.; Chinain, M.; Darius, H.T.; Cruchet, P.; Radwan, F.F.; Dickey, R.W.; Ramsdell, J.S. Biomonitoring of ciguatoxin exposure in mice using blood collection cards. Toxicon 2005, 46, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Dechraoui, M.-Y.B.; Wang, Z.; Ramsdell, J.S. Optimization of ciguatoxin extraction method from blood for Pacific ciguatoxin (P-CTX-1). Toxicon 2007, 49, 100–105. [Google Scholar] [CrossRef]
- Pauillac, S.; Branaa, P.; Chinain, M.; Naar, J. The reversed micellar medium as a universal tool for the development of antibody-based assays to marine phycotoxins using small amount of toxic material. In Harmful Algal Bloom; Hallegraeff, G.M., Blackburn, S.I., Bolch, C.J., Lewis, R.J., Eds.; Intergovernmental Oceanographic Commission of UNESCO: Hobart, Tasmania, 2001; pp. 288–291. [Google Scholar]
- Tsumuraya, T.; Fujii, I.; Inoue, M.; Tatami, A.; Miyazaki, K.; Hirama, M. Production of monoclonal antibodies for sandwich immunoassay detection of ciguatoxin 51-hydroxyCTX3C. Toxicon 2006, 48, 287–294. [Google Scholar] [CrossRef]
- Campora, C.E.; Hokama, Y.; Ebesu, J.S. Comparative analysis of purified Pacific and Caribbean ciguatoxin congeners and related marine toxins using a modified elisa technique. J. Clin. Lab. Anal. 2006, 20, 121–125. [Google Scholar] [CrossRef]
- Fairey, E.R.; Edmunds, J.G.; Ramsdell, J.S. A Cell-Based Assay for Brevetoxins, Saxitoxins, and Ciguatoxins Using a Stably Expressed c-fos–Luciferase Reporter Gene. Anal. Biochem. 1997, 251, 129–132. [Google Scholar] [CrossRef]
- Fairey, E.R.; Ramsdell, J.S. Reporter gene assays for algal-derived toxins. Nat. Toxins 1999, 7, 415–421. [Google Scholar] [CrossRef]
- A Catterall, W. Neurotoxins that Act on Voltage-Sensitive Sodium Channels in Excitable Membranes. Annu. Rev. Pharmacol. Toxicol. 1980, 20, 15–43. [Google Scholar] [CrossRef]
- Wang, G.K. Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell. Signal. 2003, 15, 151–159. [Google Scholar] [CrossRef]
- Leão-Martins, J.M.; Lozano-Leon, A.; Giraldez, J.; Vilariño, O.; Gago-Martínez, A. Preliminary Results on the Evaluation of the Occurrence of Tetrodotoxin Associated to Marine Vibrio spp. in Bivalves from the Galician Rias (Northwest of Spain). Mar. Drugs 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Kogure, K.; Tamplin, M.L.; Simidu, U.; Colwell, R.R. A tissue culture assay for tetrodotoxin, saxitoxin and related toxins. Toxicon 1988, 26, 191–197. [Google Scholar] [CrossRef]
- Caillaud, A.; Eixarch, H.; De La Iglesia, P.; Rodríguez, M.; Domínguez, L.; Andree, K.; Diogène, J. Towards the standardisation of the neuroblastoma (neuro-2a) cell-based assay for ciguatoxin-like toxicity detection in fish: Application to fish caught in the Canary Islands. Food Addit. Contam. Part A 2012, 29, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Bienfang, P.; Oben, B.; DeFelice, S.; Moeller, P.; Huncik, K.; Oben, P.; Toonen, R.; Daly-Engel, T.; Bowen, B. Ciguatera: The detection of neurotoxins in carnivorous reef fish from the coast of Cameroon, West Africa. Afr. J. Mar. Sci. 2008, 30, 533–540. [Google Scholar] [CrossRef]
- Campora, C.E.; Dierking, J.; Tamaru, C.S.; Hokama, Y.; Vincent, D. Detection of ciguatoxin in fish tissue using sandwich ELISA and neuroblastoma cell bioassay. J. Clin. Lab. Anal. 2008, 22, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Dickey, R.W. Ciguatera toxins: Chemistry, toxicology and detection. In Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection; Botana, L.M., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2008; pp. 479–500. [Google Scholar]
- Estevez, P.; Castro, D.; Leão-Martins, J.M.; Yasumoto, T.; Dickey, R.; Gago-Martinez, A. Implementation of liquid chromatography tandem mass spectrometry for the analysis of ciguatera fish poisoning in contaminated fish samples from Atlantic coasts. Food Chem. 2019, 280, 8–14. [Google Scholar] [CrossRef]
- Caillaud, A.; De La Iglesia, P.; Darius, H.T.; Pauillac, S.; Aligizaki, K.; Fraga, S.; Chinain, M.; Diogène, J. Update on Methodologies Available for Ciguatoxin Determination: Perspectives to Confront the Onset of Ciguatera Fish Poisoning in Europe [1]. Mar. Drugs 2010, 8, 1838–1907. [Google Scholar] [CrossRef]
- Pawlowiez, R.; Darius, H.T.; Cruchet, P.; Rossi, F.; Caillaud, A.; Laurent, D.; Chinain, M. Evaluation of seafood toxicity in the Australes archipelago (French Polynesia) using the neuroblastoma cell-based assay. Food Addit. Contam. Part A 2013, 30, 567–586. [Google Scholar] [CrossRef]
- Aballay-Gonzalez, A.; Ulloa, V.; Rivera, A.; Hernandez, V.; Silva, M.; Caprile, T.; Delgado-Rivera, L.; Astuya, A. Matrix effects on a cell-based assay used for the detection of paralytic shellfish toxins in bivalve shellfish samples. Food Addit. Contam. Part A 2016, 33, 869–875. [Google Scholar] [CrossRef]
- Wong, C.K.; Hung, P.; Lee, K.L.; Kam, K.M. Solid-phase extraction clean-up of ciguatoxin-contaminated coral fish extracts for use in the mouse bioassay. Food Addit. Contam. Part A 2009, 26, 236–247. [Google Scholar] [CrossRef]
- Loeffler, C.; Robertson, A.; Quintana, H.A.F.; Silander, M.C.; Smith, T.B.; Olsen, D. Ciguatoxin prevalence in 4 commercial fish species along an oceanic exposure gradient in the US Virgin Islands. Environ. Toxicol. Chem. 2018, 37, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Moreiras, G.; Leão, J.M.; Gago-Martínez, A. Design of experiments for the optimization of electrospray ionization in the LC-MS/MS analysis of ciguatoxins. J. Mass Spectrom. 2018, 53, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Dechraoui, M.-Y.B.; Tiedeken, J.A.; Persad, R.; Wang, Z.; Granade, H.R.; Dickey, R.W.; Ramsdell, J.S. Use of two detection methods to discriminate ciguatoxins from brevetoxins: Application to great barracuda from Florida Keys. Toxicon 2005, 46, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Hoy, A.W.W.; Sellin, M. Ciguatera and mannitol: In Vivo and In Vitro assessment in mice. Toxicon 1993, 31, 1039–1050. [Google Scholar] [CrossRef]
- Lewis, R.J.; Holmes, M.J. Origin and transfer of toxins involved in ciguatera. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1993, 106, 615–628. [Google Scholar] [CrossRef]
- Bidard, J.N.; Vijverberg, H.P.; Frelin, C.; Chungue, E.; Legrand, A.M.; Bagnis, R.; Lazdunski, M. Ciguatoxin is a novel type of NaC channel toxin. J. Biol. Chem. 1984, 259, 8353–8357. [Google Scholar]
- Lepage, K.T.; Dickey, R.W.; Gerwick, W.H.; Jester, E.L.; Murray, T.F. On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies. Crit. Rev. Neurobiol. 2005, 17, 27–50. [Google Scholar] [CrossRef]
- Wu, J.J.; Mak, Y.L.; Murphy, M.B.; Lam, J.C.W.; Chan, W.H.; Wang, M.; Chan, L.L.; Lam, P.K.S. Validation of an accelerated solvent extraction liquid chromatography–tandem mass spectrometry method for Pacific ciguatoxin-1 in fish flesh and comparison with the mouse neuroblastoma assay. Anal. Bioanal. Chem. 2011, 400, 3165–3175. [Google Scholar] [CrossRef]
- Pöch, G.; Pancheva, S.N. Calculating slope and ED50 of additive dose-response curves, and application of these tabulated parameter values. J. Pharmacol. Toxicol. Methods 1995, 33, 137–145. [Google Scholar] [CrossRef]
- Yogi, K.; Oshiro, N.; Inafuku, Y.; Hirama, M.; Yasumoto, T. Detailed LC-MS/MS Analysis of Ciguatoxins Revealing Distinct Regional and Species Characteristics in Fish and Causative Alga from the Pacific. Anal. Chem. 2011, 83, 8886–8891. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID Sample | Species | Common Name | Total Lipids* (g/100g) |
|---|---|---|---|
| NC1 | Pagrus pagrus | Red porgy | <3% (0.7 g/100g) |
| NC2 | Epinephelus marginatus | Dusky grouper | <3% (1.0 g/100g) |
| NC3 | Pomatomus saltatrix | Bluefish | 3–6% (4.2 g/100g) |
| NC4 | Seriola dumerili | Greater amberjack | 3–6% (5.2 g/100g) |
| NC5 | Acanthocybium solandri | Wahoo | >6% (9.4 g/100g) |
| CTX Analogue | IC50 ± σ (pg·well−1) | /Hill Slope/ |
|---|---|---|
| CTX1B | 0.26 ± 0.07 | 2.3 ± 0.48 |
| CTX3C | 0.43 ± 0.06 | 2.1 ± 0.49 |
| C-CTX1 | 0.44 ± 0.07 | 2.4 ± 0.44 |
| CTXs Type | Analysis Conditions | IC50 ± SD (pg∙well−1) | ng CTXs∙g−1 Fish ± SD | /Hill slope/ ± SD | DR ± SD | DRcorr ± SD | IC50(corr) ± SD | ng CTXs∙g−1 Fishcorr ± SD |
|---|---|---|---|---|---|---|---|---|
| CCTX1 | No matrix | 0.48 ± 0.04 | 0.024 ± 0.002 | 3.5 ± 0.5 | - | - | - | 0.024 ± 0.002 |
| Purified | 0.56 ± 0.04 | 0.027 ± 0.002 | 1.6 ± 0.1 | 1.1 ± 0.1 | 1.3 ± 0.1 | 0.6 ± 0.1 | 0.031 ± 0.003 | |
| Unpurified | 1.3 ± 0.1 | 0.06 ± 0.01 | 1.5 ± 0.3 | 2.6 ± 0.2 | 3.6 ± 0.9 | 1.8 ± 0.2 | 0.12 ± 0.02 | |
| CTX1B | No matrix | 0.26 ± 0.02 | 0.013 ± 0.001 | 2.4 ± 0.1 | - | - | - | 0.013 ± 0.001 |
| Purified | 0.27 ± 0.02 | 0.013 ± 0.001 | 2.4 ± 0.2 | 1.0 ± 0.1 | 1.0 ± 0.2 | 0.27 ± 0.05 | 0.014 ± 0.003 | |
| Unpurified | 0.34 ± 0.02 | 0.017 ± 0.001 | 2.3 ± 0.4 | 1.3 ± 0.1 | 1.6 ± 0.3 | 0.4 ± 0.1 | 0.022 ± 0.006 |
| CTXs Type | Analysis Conditions | LOD | LOQ | ||
|---|---|---|---|---|---|
| IC10 ± SD (pg∙well−1) | ng CTXs∙g−1 Fish ± SD | IC20 ± SD (pg∙well−1) | ng CTXs∙g−1 Fish ± SD | ||
| CCTX1 | No matrix | 0.20 ± 0.02 | 0.010 ± 0,002 | 0.29 ± 0.03 | 0.014 ± 0.002 |
| Purified | 0.12 ± 0.09 | 0.008 ± 0,001 | 0.23 ± 0.02 | 0.012 ± 0.001 | |
| Unpurified | 0.26 ± 0.02 | 0.013 ± 0,004 | 0.46 ± 0.09 | 0.023 ± 0.005 | |
| CTX1B | No matrix | 0.12 ± 0.02 | 0.007 ± 9 × 10−3 | 0.17 ± 0.02 | 0.009 ± 9 × 10−3 |
| Purified | 0.10 ± 0.01 | 0.007 ± 6 × 10−3 | 0.14 ± 0.01 | 0.008 ± 3 × 10−3 | |
| Unpurified | 0.13 ± 0.01 | 0.005 ± 5 × 10−3 | 0.19 ± 0.01 | 0.007 ± 5 × 10−3 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, D.; Manger, R.; Vilariño, O.; Gago-Martínez, A. Evaluation of Matrix Issues in the Applicability of the Neuro-2a Cell Based Assay on the Detection of CTX in Fish Samples. Toxins 2020, 12, 308. https://doi.org/10.3390/toxins12050308
Castro D, Manger R, Vilariño O, Gago-Martínez A. Evaluation of Matrix Issues in the Applicability of the Neuro-2a Cell Based Assay on the Detection of CTX in Fish Samples. Toxins. 2020; 12(5):308. https://doi.org/10.3390/toxins12050308
Chicago/Turabian StyleCastro, David, Ronald Manger, Oscar Vilariño, and Ana Gago-Martínez. 2020. "Evaluation of Matrix Issues in the Applicability of the Neuro-2a Cell Based Assay on the Detection of CTX in Fish Samples" Toxins 12, no. 5: 308. https://doi.org/10.3390/toxins12050308
APA StyleCastro, D., Manger, R., Vilariño, O., & Gago-Martínez, A. (2020). Evaluation of Matrix Issues in the Applicability of the Neuro-2a Cell Based Assay on the Detection of CTX in Fish Samples. Toxins, 12(5), 308. https://doi.org/10.3390/toxins12050308