Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis

 ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Upregulation of ESM1 in Unilateral Ureteral Obstruction (UUO)
2.2. Overexpression of ESM1 in Mouse MES 13 Cells
2.3. Overexpression of ESM1 Had No Effect on the Growth of MES 13 Cells
2.4. Overexpression of ESM1 Increased Motility and Migration of MES 13 Cells
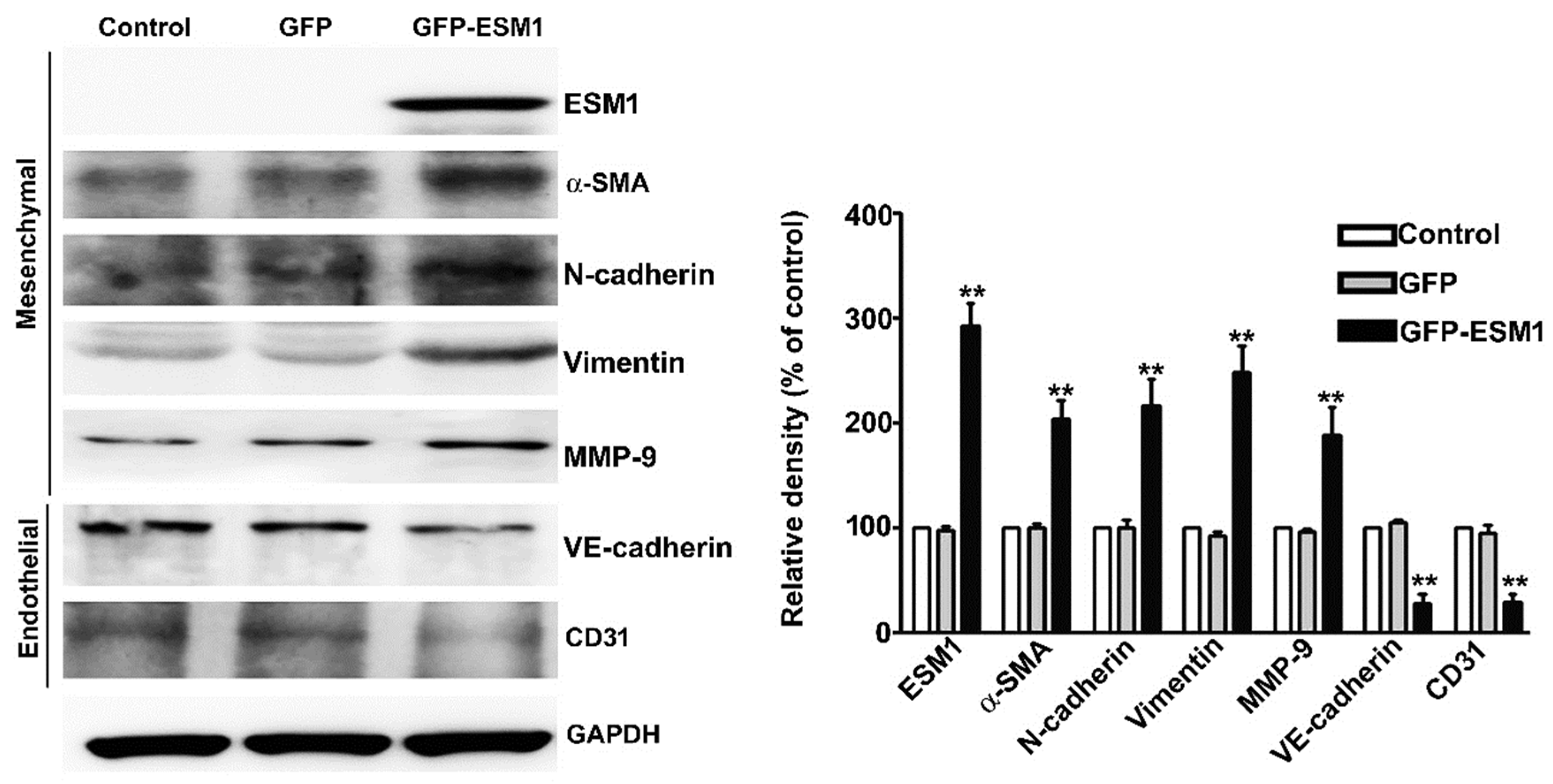
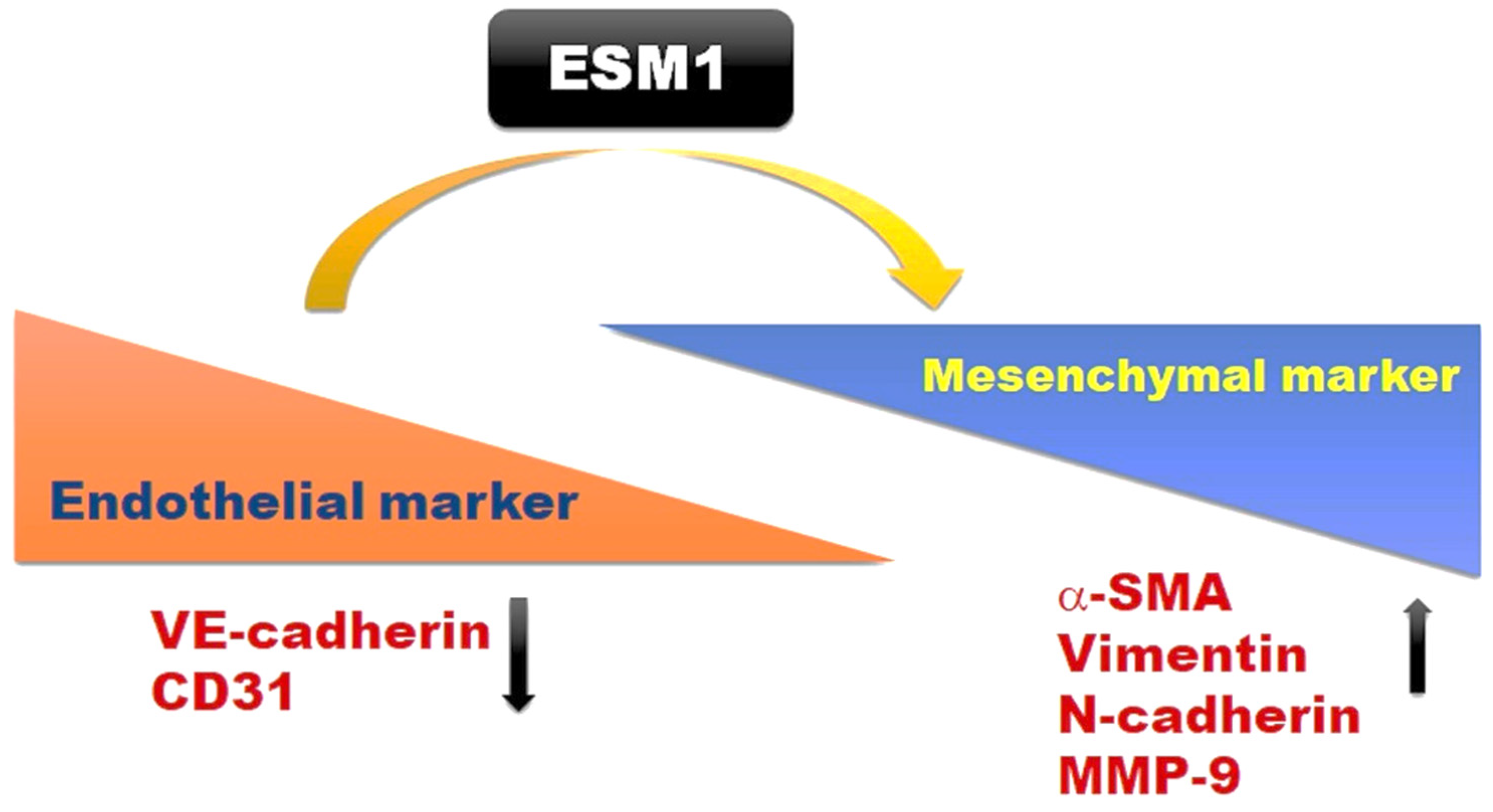
2.5. Overexpression of ESM1 Induced EndoMT in MES 13 Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemical Reagent
5.2. Cell Culture
5.3. Gene Transfection
5.4. Cell Viability Assay
5.5. Cell Proliferation Assay
5.6. Wound Healing Assay
5.7. Cell Migration Assay
5.8. Western Blot Analysis
5.9. UUO Mouse Model
5.10. Immunohistochemical Analysis and Masson’s Trichrome Staining
5.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Prim. 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appenroth, D.; Lupp, A.; Kriegsmann, J.; Sawall, S.; Splinther, J.; Sommer, M.; Stein, G.; Fleck, C. Temporary warm ischaemia, 5/6 nephrectomy and single uranylnitrate administration—Comparison of three models intended to cause renal fibrosis in rats. Exp. Toxicol. Pathol. 2001, 53, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Adam, E.; Lyon, M.; Depontieu, F.; Motte, V.; Landolfi, C.; Lortat-Jacob, H.; Bechard, D.; Lassalle, P.; Delehedde, M. Endocan or endothelial cell specific molecule-1 (ESM-1): A potential novel endothelial cell marker and a new target for cancer therapy. Biochim. Biophys. Acta 2006, 1765, 25–37. [Google Scholar] [CrossRef]
- Leroy, X.; Aubert, S.; Zini, L.; Franquet, H.; Kervoaze, G.; Villers, A.; Delehedde, M.; Copin, M.C.; Lassalle, P. Vascular endocan (ESM-1) is markedly overexpressed in clear cell renal cell carcinoma. Histopathology 2010, 56, 180–187. [Google Scholar] [CrossRef]
- Balta, S.; Mikhailidis, D.P.; Demirkol, S.; Ozturk, C.; Celik, T.; Iyisoy, A. Endocan: A novel inflammatory indicator in cardiovascular disease? Atherosclerosis 2015, 243, 339–343. [Google Scholar] [CrossRef]
- Kose, M.; Emet, S.; Akpinar, T.S.; Kocaaga, M.; Cakmak, R.; Akarsu, M.; Yuruyen, G.; Arman, Y.; Tukek, T. Serum Endocan Level and the Severity of Coronary Artery Disease: A Pilot Study. Angiology 2015, 66, 727–731. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Siriopol, D.; Saglam, M.; Kurt, Y.G.; Unal, H.U.; Eyileten, T.; Gok, M.; Cetinkaya, H.; Oguz, Y.; Sari, S.; et al. Plasma endocan levels associate with inflammation, vascular abnormalities, cardiovascular events, and survival in chronic kidney disease. Kidney Int. 2014, 86, 1213–1220. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Kim, J.S.; Kim, S.Y.; Kim, Y.G.; Moon, J.Y.; Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Lee, S.H. Plasma endocan level and prognosis of immunoglobulin A nephropathy. Kidney Res. Clin. Pract. 2016, 35, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauly, D.; Hamed, S.; Behnes, M.; Lepiorz, D.; Lang, S.; Akin, I.; Borggrefe, M.; Bertsch, T.; Hoffmann, U. Endothelial cell-specific molecule-1/endocan: Diagnostic and prognostic value in patients suffering from severe sepsis and septic shock. J. Crit. Care 2016, 31, 68–75. [Google Scholar] [CrossRef]
- Bernatsky, S.; Joseph, L.; Pineau, C.A.; Belisle, P.; Hudson, M.; Clarke, A.E. Scleroderma prevalence: Demographic variations in a population-based sample. Arthritis Rheum. 2009, 61, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balanescu, P.; Ladaru, A.; Balanescu, E.; Voiosu, T.; Baicus, C.; Dan, G.A. Endocan, Novel Potential Biomarker for Systemic Sclerosis: Results of a Pilot Study. J. Clin. Lab. Anal. 2016, 30, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Malyszko, J.; Koc-Zorawska, E.; Malyszko, J.S. Endocan Concentration in Kidney Transplant Recipients. Transplant. Proc. 2018, 50, 1798–1801. [Google Scholar] [CrossRef]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef] [Green Version]
- Satchell, S.C. The glomerular endothelium emerges as a key player in diabetic nephropathy. Kidney Int. 2012, 82, 949–951. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Tanabe, K.; Croker, B.P.; Johnson, R.J.; Grant, M.B.; Kosugi, T.; Li, Q. Endothelial dysfunction as a potential contributor in diabetic nephropathy. Nat. Rev. Nephrol. 2011, 7, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Bertram, J.F. Review: Endothelial-myofibroblast transition, a new player in diabetic renal fibrosis. Nephrology 2010, 15, 507–512. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balta, I.; Balta, S.; Demirkol, S.; Mikhailidis, D.P.; Celik, T.; Akhan, M.; Kurt, O.; Kurt, Y.G.; Aydin, I.; Kilic, S. Elevated serum levels of endocan in patients with psoriasis vulgaris: Correlations with cardiovascular risk and activity of disease. Br. J. Dermatol. 2013, 169, 1066–1070. [Google Scholar] [CrossRef]
- Scherpereel, A.; Depontieu, F.; Grigoriu, B.; Cavestri, B.; Tsicopoulos, A.; Gentina, T.; Jourdain, M.; Pugin, J.; Tonnel, A.B.; Lassalle, P. Endocan, a new endothelial marker in human sepsis. Crit. Care Med. 2006, 34, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Balta, S.; Mikhailidis, D.P.; Demirkol, S.; Ozturk, C.; Kurtoglu, E.; Demir, M.; Celik, T.; Turker, T.; Iyisoy, A. Endocan—A novel inflammatory indicator in newly diagnosed patients with hypertension: A pilot study. Angiology 2014, 65, 773–777. [Google Scholar] [CrossRef]
- Su, Y.H.; Shu, K.H.; Hu, C.P.; Cheng, C.H.; Wu, M.J.; Yu, T.M.; Chuang, Y.W.; Huang, S.T.; Chen, C.H. Serum Endocan correlated with stage of chronic kidney disease and deterioration in renal transplant recipients. Transplant. Proc. 2014, 46, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Samouilidou, E.; Bountou, E.; Papandroulaki, F.; Papamanolis, M.; Papakostas, D.; Grapsa, E. Serum Endocan Levels are Associated With Paraoxonase 1 Concentration in Patients With Chronic Kidney Disease. Ther. Apher. Dial. 2018, 22, 325–331. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, Y.A.; Xiao, H.; Wang, Y.; Wu, Y.; Yan, Y.; Zhu, Z.; Ni, M.; Pi, C.X.; Liu, M.Y.; et al. Serum endocan and circadian heart rate variability in non-dialysis stage 5 chronic kidney disease patients. Int. Urol. Nephrol. 2018, 50, 2061–2066. [Google Scholar] [CrossRef]
- Toshikuni, N.; Ozaki, K.; George, J.; Tsutsumi, M. Serum endocan as a survival predictor for patients with liver cirrhosis. Can. J. Gastroenterol. Hepatol. 2015, 29, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Voiosu, A.M.; Balanescu, P.; Daha, I.; Smarandache, B.; Radoi, A.; Mateescu, R.B.; Baicus, C.R.; Voiosu, T.A. The diagnostic and prognostic value of serum endocan in patients with cirrhotic cardiomyopathy. Rom. J. Intern. Med. 2018, 56, 182–192. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.; Ku, S.K.; Kim, S.W.; Bae, J.S. Endocan elicits severe vascular inflammatory responses in vitro and in vivo. J. Cell. Physiol. 2014, 229, 620–630. [Google Scholar] [CrossRef]
- Yang, Z.; He, L.J.; Sun, S.R. Role of Endothelial Cells in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; van Zonneveld, A.J.; ten Dijke, P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis? Trends Cardiovasc. Med. 2008, 18, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Wang, N.; Zhang, T.C. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life 2012, 64, 717–723. [Google Scholar] [CrossRef]
- Afsar, B.; Takir, M.; Kostek, O.; Covic, A.; Kanbay, M. Endocan: A new molecule playing a role in the development of hypertension and chronic kidney disease? J. Clin. Hypertens. 2014, 16, 914–916. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Wang, C.; Wang, Q.; Yang, H.; Liang, P.; Jin, F. Detection on dynamic changes of endothelial cell specific molecule-1 in acute rejection after renal transplantation. Urology 2012, 80, 738.e1–738.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, Z.; Tao, J.; Wang, J.; Liu, X.; Zhou, W.; Xu, Z.; Zhao, C.; Wang, Z.; Tan, R.; et al. Role of endothelial-to-mesenchymal transition induced by TGF-beta1 in transplant kidney interstitial fibrosis. J. Cell. Mol. Med. 2017, 21, 2359–2369. [Google Scholar] [CrossRef] [Green Version]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-beta-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Sano, Y.; Harada, J.; Tashiro, S.; Gotoh-Mandeville, R.; Maekawa, T.; Ishii, S. ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-beta signaling. J. Biol. Chem. 1999, 274, 8949–8957. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, A.; Colletti, M.; Cozzolino, A.M.; Steindler, C.; Lunadei, M.; Mancone, C.; Tripodi, M. ERK5/MAPK is activated by TGFbeta in hepatocytes and required for the GSK-3beta-mediated Snail protein stabilization. Cell. Signal. 2008, 20, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Lin, C.L.; Chiou, H.L.; Hsieh, S.C.; Lin, C.L.; Cheng, C.W.; Hung, C.H.; Tsai, J.P.; Hsieh, Y.H. Loss of endothelial cell-specific molecule 1 promotes the tumorigenicity and metastasis of prostate cancer cells through regulation of the TIMP-1/MMP-9 expression. Oncotarget 2017, 8, 13886–13897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.H.; Yang, J.S.; Lin, R.C.; Hsieh, Y.H.; Yang, S.F.; Chang, H.R.; Lu, K.H. Tomatidine Represses Invasion and Migration of Human Osteosarcoma U2OS and HOS Cells by Suppression of Presenilin 1 and c-Raf-MEK-ERK Pathway. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.S.; Hung, T.W.; Su, S.C.; Lin, C.L.; Yang, S.F.; Lee, C.C.; Yeh, C.F.; Hsieh, Y.H.; Tsai, J.P. MTA2 as a Potential Biomarker and Its Involvement in Metastatic Progression of Human Renal Cancer by miR-133b Targeting MMP-9. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.C.; Huang, C.C.; Lu, Y.T.; Yeh, C.M.; Ho, Y.T.; Yang, S.F.; Hsin, C.H.; Lin, C.W. Epigallocatechin-3-gallate inhibits migration of human nasopharyngeal carcinoma cells by repressing MMP-2 expression. J. Cell. Physiol. 2019, 234, 20915–20924. [Google Scholar] [CrossRef]
- Chien, H.J.; Ying, T.H.; Hsieh, S.C.; Lin, C.L.; Yu, Y.L.; Kao, S.H.; Hsieh, Y.H. alpha-Mangostin attenuates stemness and enhances cisplatin-induced cell death in cervical cancer stem-like cells through induction of mitochondrial-mediated apoptosis. J. Cell. Physiol. 2020, 235, 5590–5601. [Google Scholar] [CrossRef]
- Chung, S.; Kim, S.; Son, M.; Kim, M.; Koh, E.S.; Shin, S.J.; Park, C.W.; Kim, H.S. Inhibition of p300/CBP-Associated Factor Attenuates Renal Tubulointerstitial Fibrosis through Modulation of NF-kB and Nrf2. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, T.-W.; Chu, C.-Y.; Yu, C.-L.; Lee, C.-C.; Hsu, L.-S.; Chen, Y.-S.; Hsieh, Y.-H.; Tsai, J.-P. Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis. Toxins 2020, 12, 506. https://doi.org/10.3390/toxins12080506
Hung T-W, Chu C-Y, Yu C-L, Lee C-C, Hsu L-S, Chen Y-S, Hsieh Y-H, Tsai J-P. Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis. Toxins. 2020; 12(8):506. https://doi.org/10.3390/toxins12080506
Chicago/Turabian StyleHung, Tung-Wei, Chao-Yang Chu, Chen-Lin Yu, Chu-Che Lee, Li-Sung Hsu, Yong-Syuan Chen, Yi-Hsien Hsieh, and Jen-Pi Tsai. 2020. "Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis" Toxins 12, no. 8: 506. https://doi.org/10.3390/toxins12080506
APA StyleHung, T. -W., Chu, C. -Y., Yu, C. -L., Lee, C. -C., Hsu, L. -S., Chen, Y. -S., Hsieh, Y. -H., & Tsai, J. -P. (2020). Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis. Toxins, 12(8), 506. https://doi.org/10.3390/toxins12080506