Germ-Free Conditions Modulate Host Purine Metabolism, Exacerbating Adenine-Induced Kidney Damage
 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
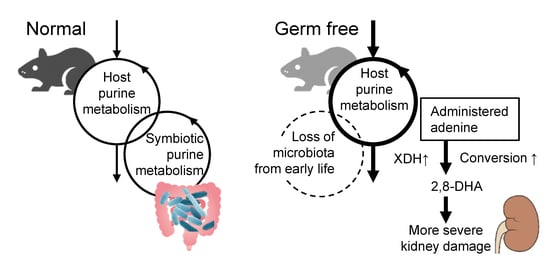
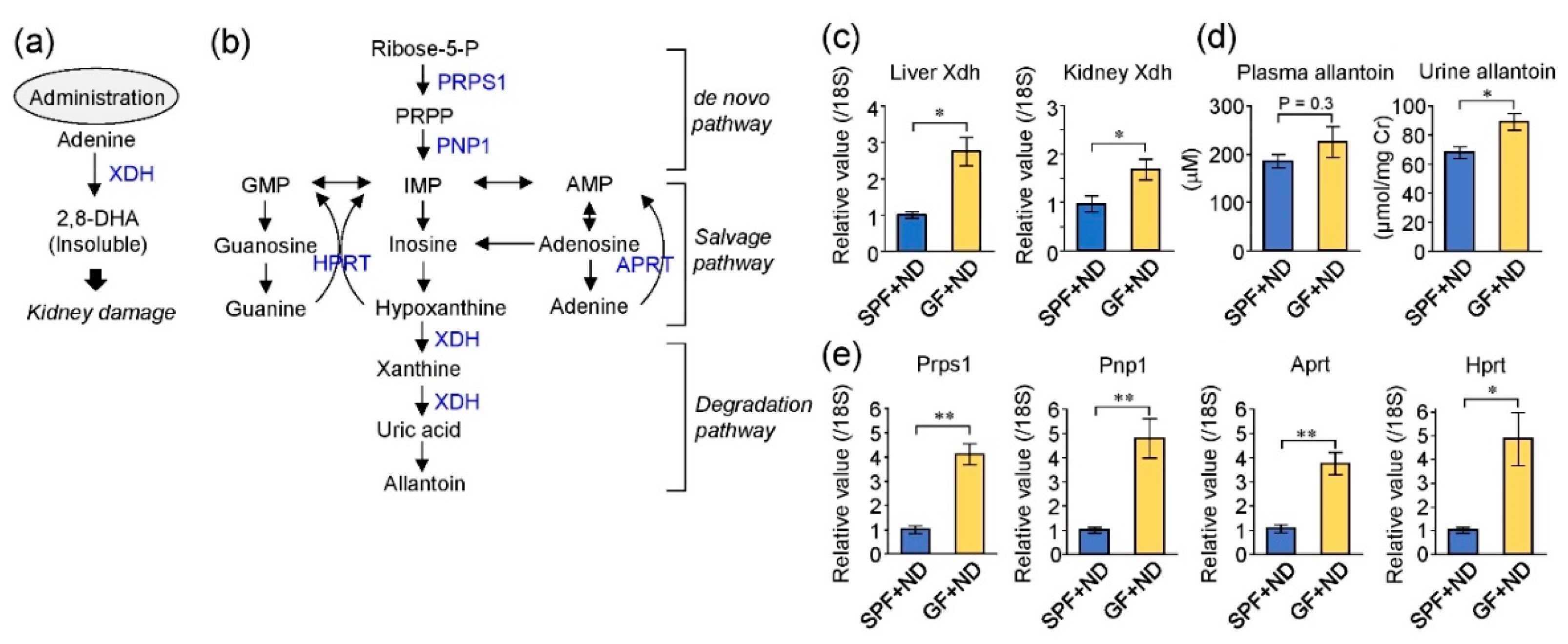
2.1. The Germ-Free Condition Increased the Expression of Purine Metabolizing Enzymes
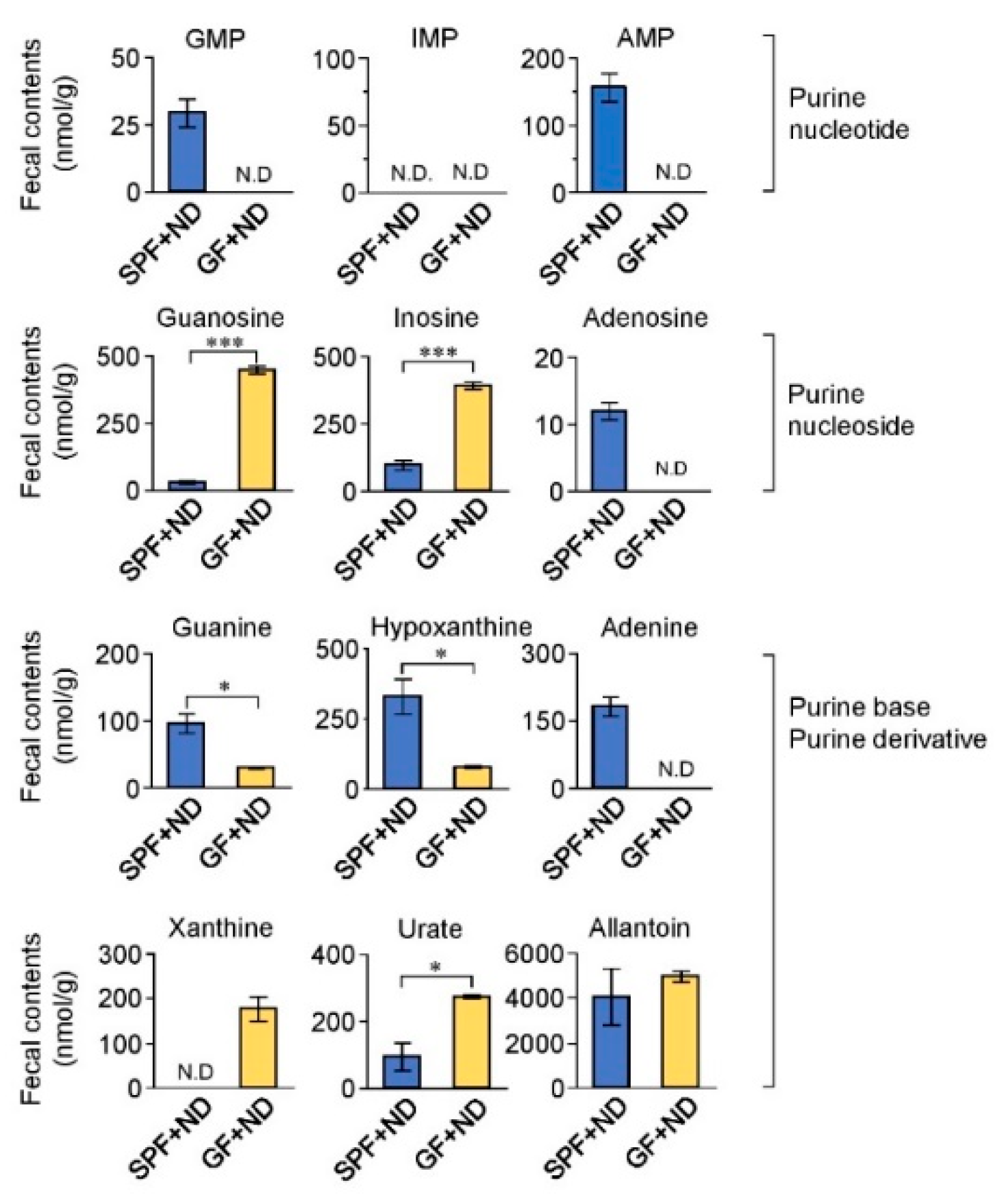
2.2. Fecal Purine Metabolites Were Altered between GF and SPF Conditions
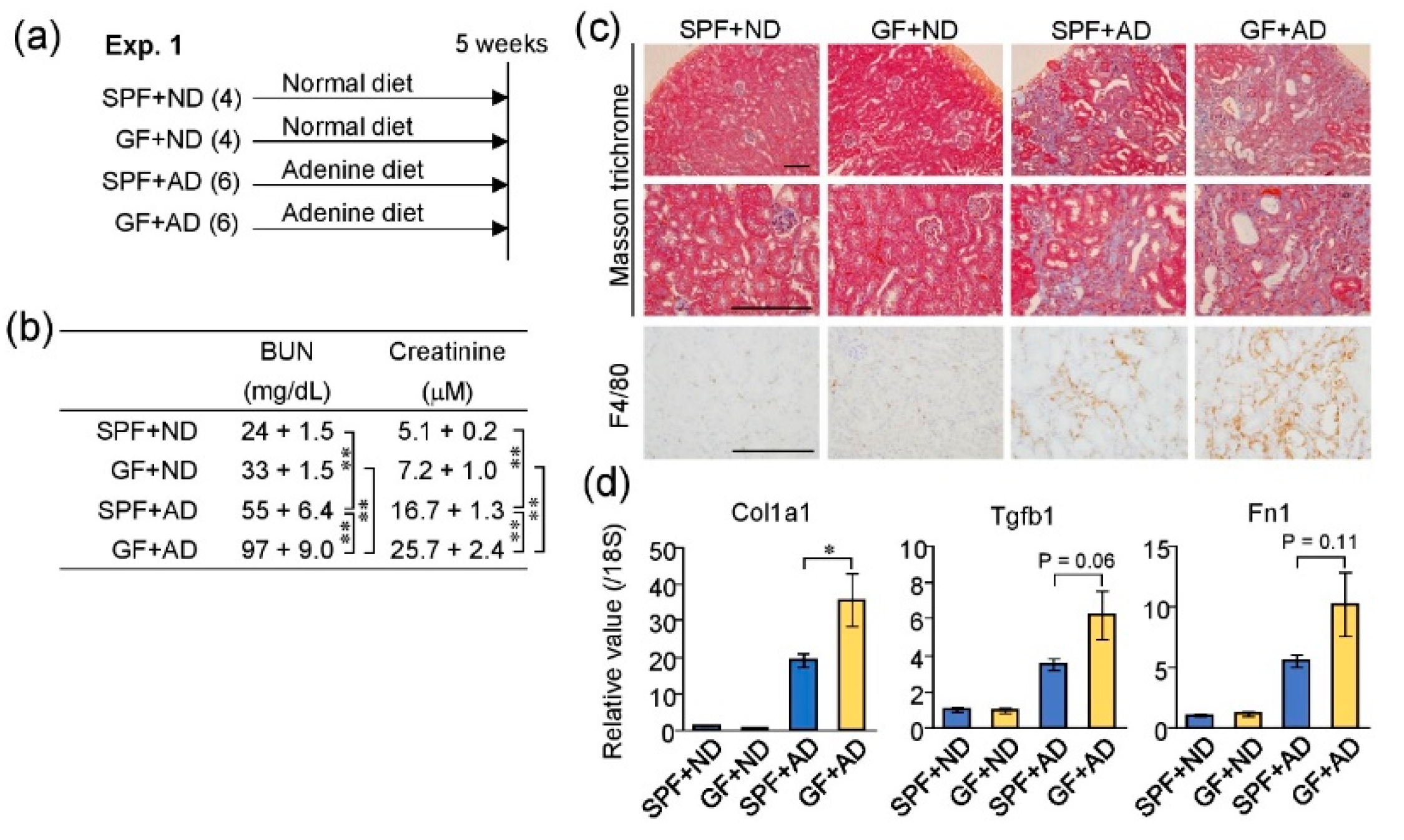
2.3. Germ-Free Conditions Exacerbated Adenine-Induced Kidney Damage in Mice
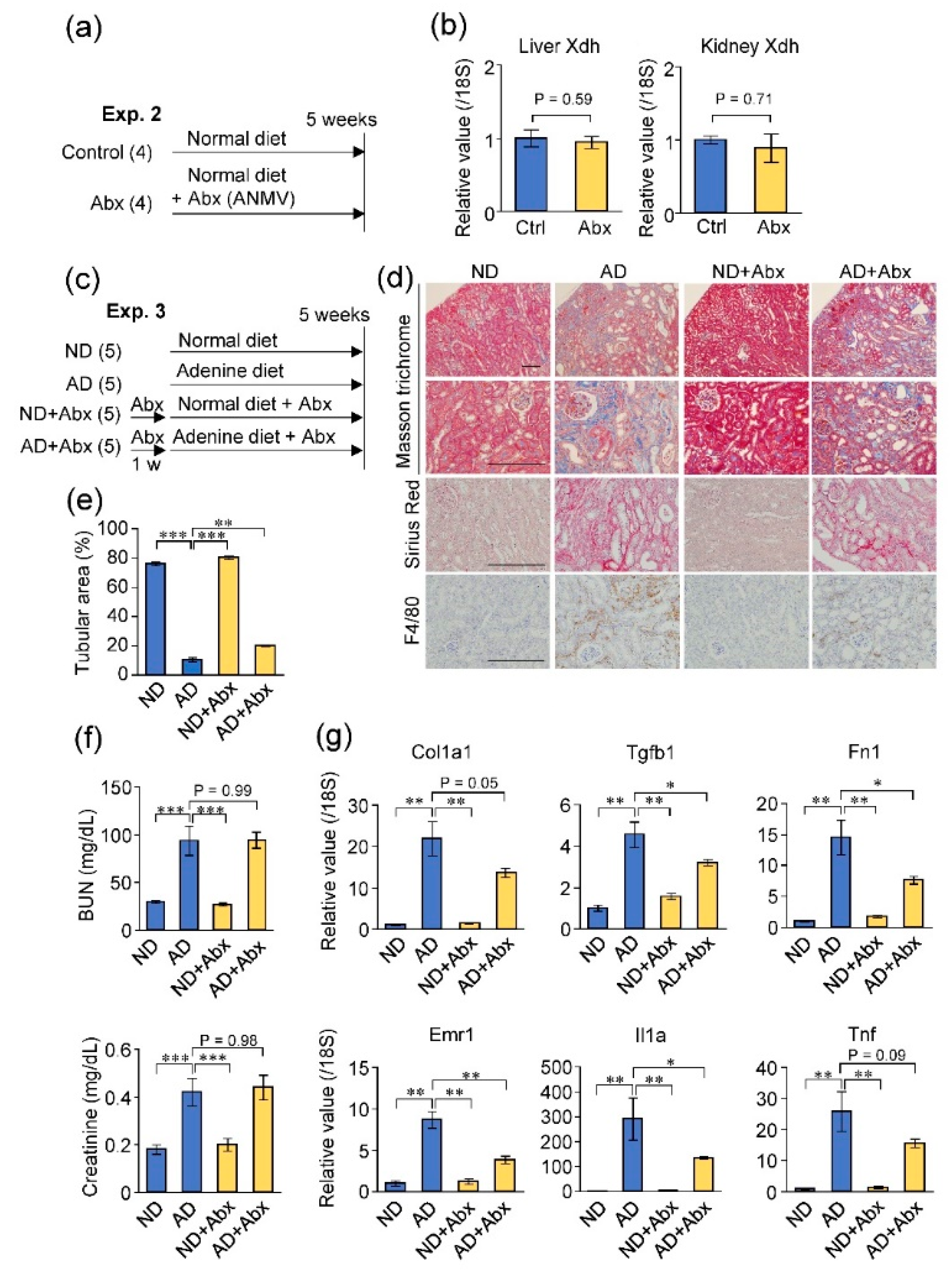
2.4. Antibiotic Treatment Did Not Exacerbate Adenine-Induced Kidney Damage
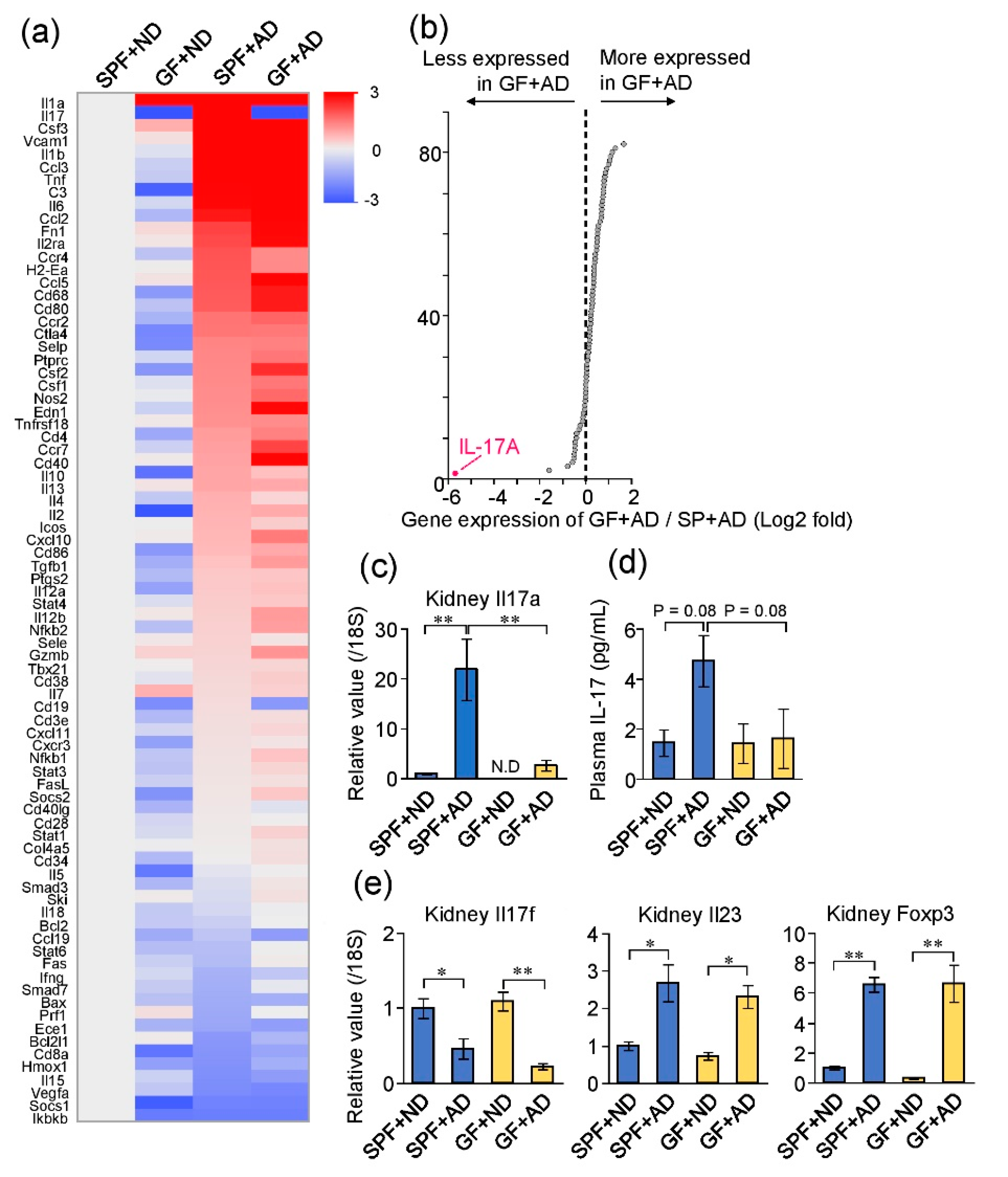
2.5. Differences in Renal Inflammatory Responses between GF and SPF Mice
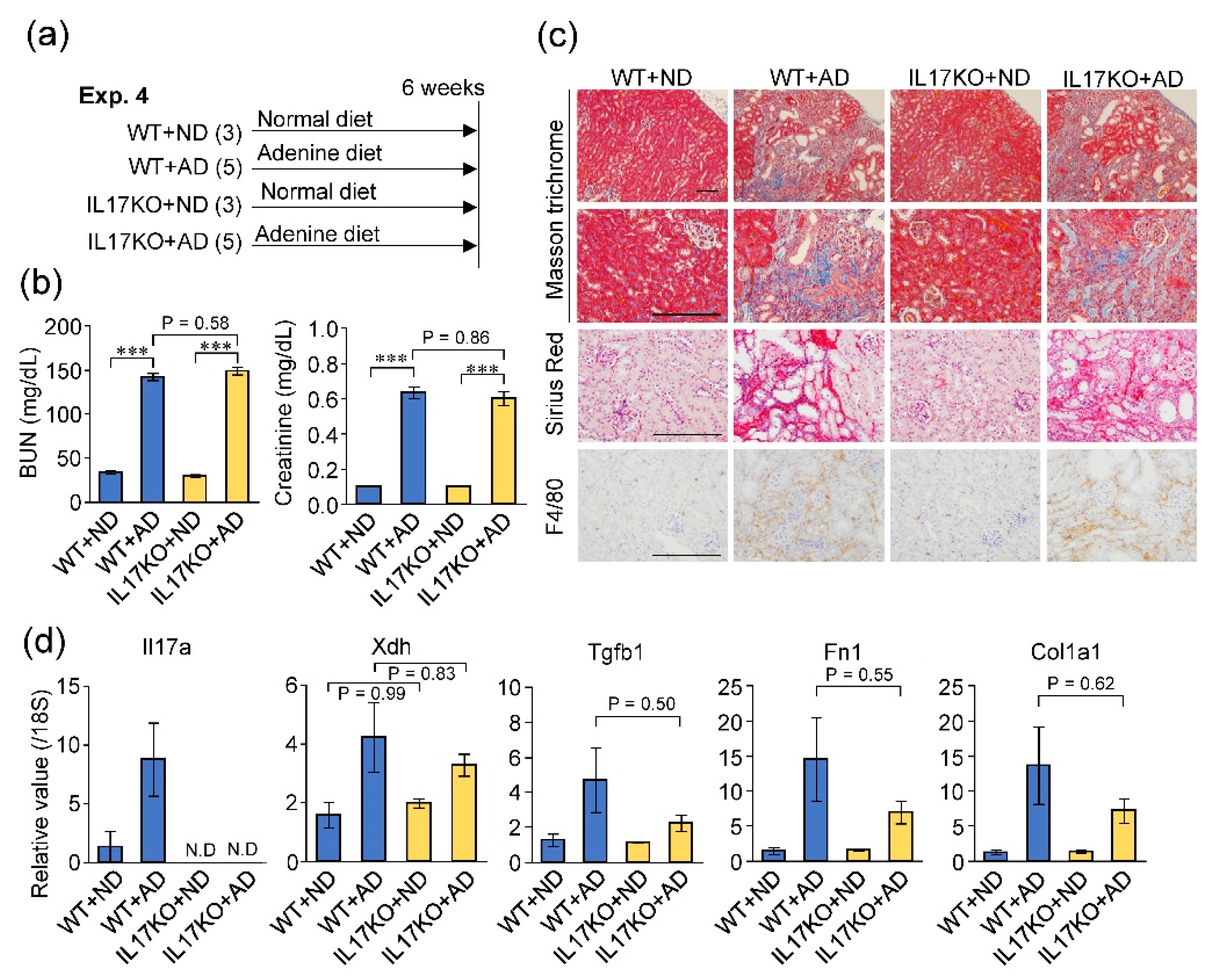
2.6. Deficiency of IL-17A Did Not Influence Adenine-Induced Kidney Damage
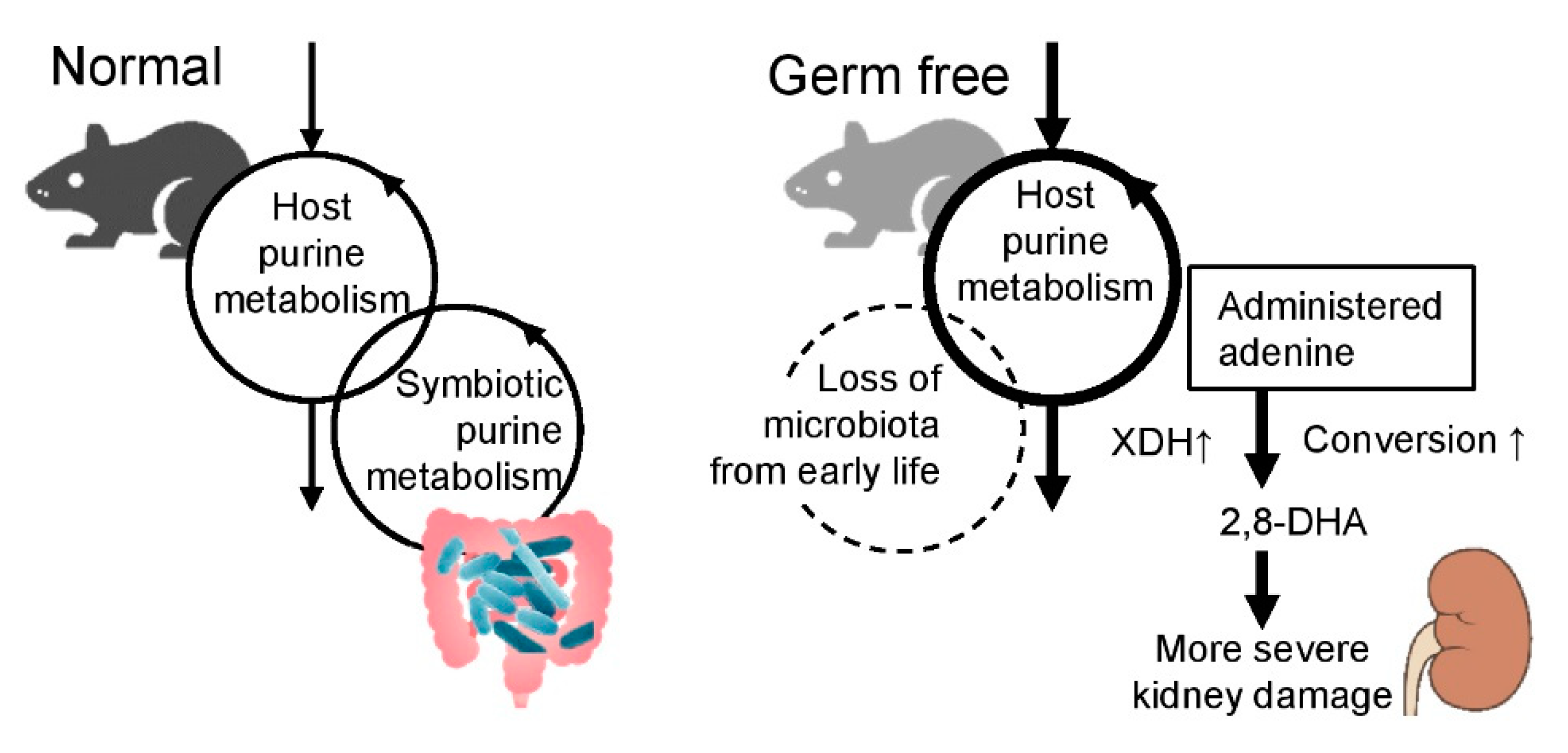
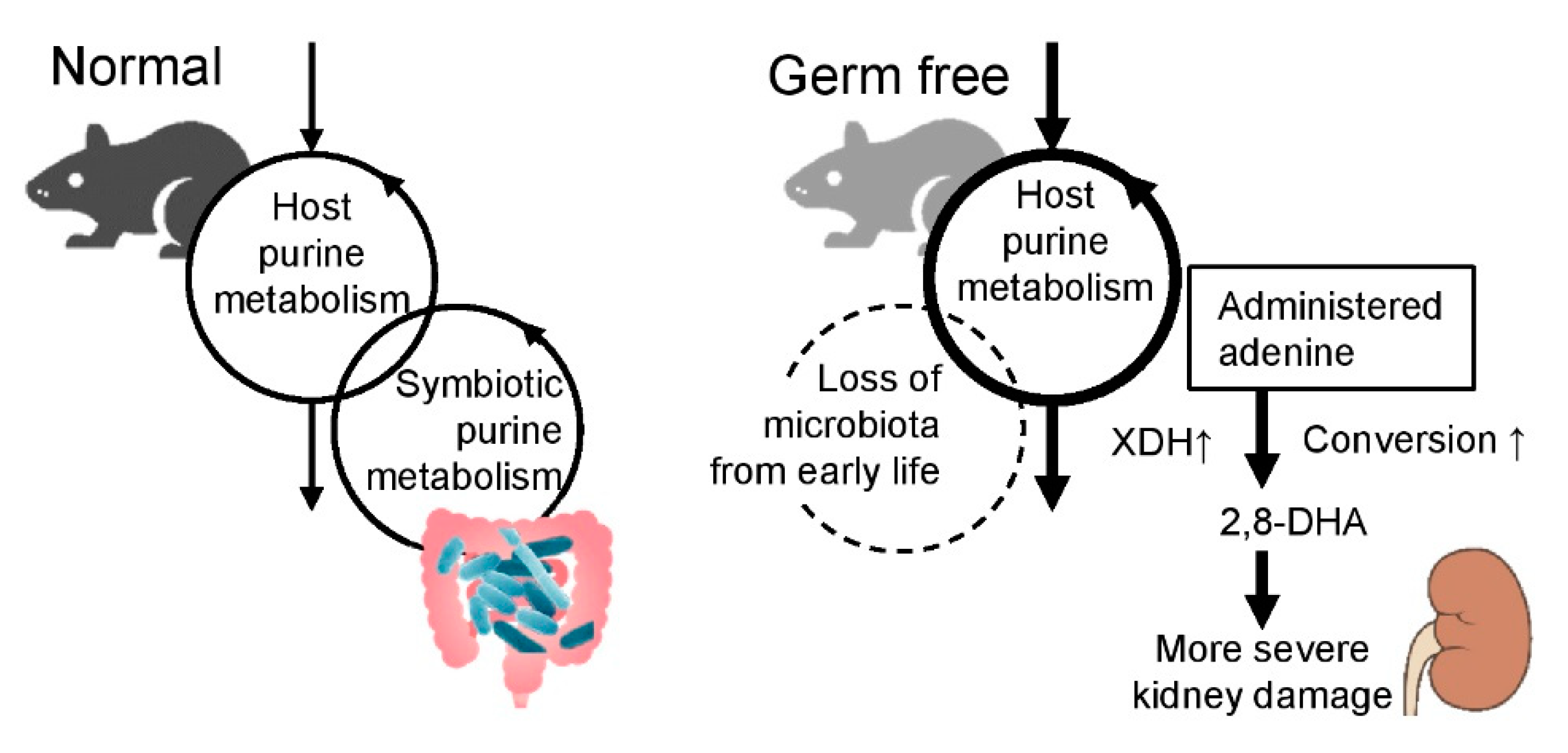
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animal Experiments
5.2. Quantitative PCR
5.3. Immune Array Analysis
5.4. Measurement of Allantoin and Purine Metabolites
5.5. Measurement of Renal Function Parameters
5.6. Measurement of Plasma IL-17
5.7. Histology
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, T.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Burtey, S. Accumulation of protein-bound uremic toxins: The kidney remains the leading culprit in the gut-liver-kidney axis. Kidney Int. 2020, 97, 1102–1104. [Google Scholar] [CrossRef]
- Rukavina Mikusic, N.L.; Kouyoumdzian, N.M.; Choi, M.R. Gut microbiota and chronic kidney disease: Evidences and mechanisms that mediate a new communication in the gastrointestinal-renal axis. Pflug. Arch. 2020, 472, 303–320. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, K.; Saigusa, D.; Kanemitsu, Y.; Matsumoto, Y.; Thanai, P.; Suzuki, N.; Mise, K.; Yamaguchi, H.; Nakamura, T.; Asaji, K.; et al. Gut microbiome-derived phenyl sulfate contributes to albuminuria in diabetic kidney disease. Nat. Commun. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Yang, J.; Kim, C.J.; Go, Y.S.; Lee, H.Y.; Kim, M.G.; Oh, S.W.; Cho, W.Y.; Im, S.H.; Jo, S.K. Intestinal microbiota controls acute kidney injury severity by immune modulation. Kidney Int. 2020. [Google Scholar] [CrossRef]
- Sueyoshi, M.; Fukunaga, M.; Mei, M.; Nakajima, A.; Tanaka, G.; Murase, T.; Narita, Y.; Hirata, S.; Kadowaki, D. Effects of lactulose on renal function and gut microbiota in adenine-induced chronic kidney disease rats. Clin. Exp. Nephrol. 2019, 23, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, K.; Wakino, S.; Irie, J.; Miyamoto, J.; Matsui, A.; Tajima, T.; Itoh, T.; Oshima, Y.; Yoshifuji, A.; Kimura, I.; et al. Contribution of uremic dysbiosis to insulin resistance and sarcopenia. Nephrol. Dial. Transpl. 2020. [Google Scholar] [CrossRef]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef]
- Evenepoel, P.; Dejongh, S.; Verbeke, K.; Meijers, B. The Role of Gut Dysbiosis in the Bone-Vascular Axis in Chronic Kidney Disease. Toxins 2020, 12. [Google Scholar] [CrossRef]
- Glorieux, G.; Gryp, T.; Perna, A. Gut-Derived Metabolites and Their Role in Immune Dysfunction in Chronic Kidney Disease. Toxins 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onal, E.M.; Afsar, B.; Covic, A.; Vaziri, N.D.; Kanbay, M. Gut microbiota and inflammation in chronic kidney disease and their roles in the development of cardiovascular disease. Hypertens. Res. 2019, 42, 123–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Oliveira, V.; Amano, M.T.; Correa-Costa, M.; Castoldi, A.; Felizardo, R.J.; de Almeida, D.C.; Bassi, E.J.; Moraes-Vieira, P.M.; Hiyane, M.I.; Rodas, A.C.; et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Chen, X.; Kwan, T.K.; Loh, Y.W.; Singer, J.; Liu, Y.; Ma, J.; Tan, J.; Macia, L.; Mackay, C.R.; et al. Dietary Fiber Protects against Diabetic Nephropathy through Short-Chain Fatty Acid-Mediated Activation of G Protein-Coupled Receptors GPR43 and GPR109A. J. Am. Soc. Nephrol. 2020, 31, 1267–1281. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Koppe, L.; Fouque, D.; Soulage, C.O. The Role of Gut Microbiota and Diet on Uremic Retention Solutes Production in the Context of Chronic Kidney Disease. Toxins 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Emal, D.; Rampanelli, E.; Stroo, I.; Butter, L.M.; Teske, G.J.; Claessen, N.; Stokman, G.; Florquin, S.; Leemans, J.C.; Dessing, M.C. Depletion of Gut Microbiota Protects against Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2017, 28, 1450–1461. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.; Kesper, M.S.; Marschner, J.A.; Konrad, L.; Ryu, M.; Kumar Vr, S.; Kulkarni, O.P.; Mulay, S.R.; Romoli, S.; Demleitner, J.; et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J. Am. Soc. Nephrol. 2017, 28, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, C.F.; Paust, H.J.; Krohn, S.; Koyro, T.; Brix, S.R.; Riedel, J.H.; Bartsch, P.; Wiech, T.; Meyer-Schwesinger, C.; Huang, J.; et al. Autoimmune Renal Disease Is Exacerbated by S1P-Receptor-1-Dependent Intestinal Th17 Cell Migration to the Kidney. Immunity 2016, 45, 1078–1092. [Google Scholar] [CrossRef] [Green Version]
- Mu, Q.; Tavella, V.J.; Kirby, J.L.; Cecere, T.E.; Chung, M.; Lee, J.; Li, S.; Ahmed, S.A.; Eden, K.; Allen, I.C.; et al. Antibiotics ameliorate lupus-like symptoms in mice. Sci. Rep. 2017, 7, 13675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.R.; Gandolfo, M.T.; Ko, G.J.; Satpute, S.; Racusen, L.; Rabb, H. Early exposure to germs modifies kidney damage and inflammation after experimental ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2009, 297, F1457–F1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanto-Hara, F.; Kanemitsu, Y.; Fukuda, S.; Kikuchi, K.; Asaji, K.; Saigusa, D.; Iwasaki, T.; Ho, H.J.; Mishima, E.; Suzuki, T.; et al. The guanylate cyclase C agonist linaclotide ameliorates the gut-cardio-renal axis in an adenine-induced mouse model of chronic kidney disease. Nephrol. Dial. Transplant. 2020, 35, 250–264. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Kanemitsu, Y.; Saigusa, D.; Mukawa, C.; Asaji, K.; Matsumoto, Y.; Tsukamoto, H.; Tachikawa, T.; Tsukimi, T.; et al. Canagliflozin reduces plasma uremic toxins and alters the intestinal microbiota composition in a chronic kidney disease mouse model. Am. J. Physiol. Ren. Physiol. 2018, 315, F824–F833. [Google Scholar] [CrossRef] [Green Version]
- Klinkhammer, B.M.; Djudjaj, S.; Kunter, U.; Palsson, R.; Edvardsson, V.O.; Wiech, T.; Thorsteinsdottir, M.; Hardarson, S.; Foresto-Neto, O.; Mulay, S.R.; et al. Cellular and Molecular Mechanisms of Kidney Injury in 2,8-Dihydroxyadenine Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 799–816. [Google Scholar] [CrossRef]
- Chiaro, T.R.; Soto, R.; Zac Stephens, W.; Kubinak, J.L.; Petersen, C.; Gogokhia, L.; Bell, R.; Delgado, J.C.; Cox, J.; Voth, W.; et al. A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Lee, J.S.; Wang, R.; Goldberg, M.; Kao, D.; Colgan, S. Microbiota-derived Purines Support Intestinal Proliferation and Mucus Barrier Integrity. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Ogawa, J.; Soong, C.L.; Kishino, S.; Li, Q.S.; Horinouchi, N.; Shimizu, S. Screening and industrial application of unique microbial reactions involved in nucleic acid and lipid metabolisms. Biosci. Biotechnol. Biochem. 2006, 70, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Kamijo-Ikemori, A.; Sugaya, T.; Hibi, C.; Nakamura, T.; Murase, T.; Oikawa, T.; Hoshino, S.; Hisamichi, M.; Hirata, K.; Kimura, K.; et al. Renoprotective effect of the xanthine oxidoreductase inhibitor topiroxostat on adenine-induced renal injury. Am. J. Physiol. Ren. Physiol. 2016, 310, F1366–F1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Doi, K.; Maeda-Mamiya, R.; Negishi, K.; Portilla, D.; Sugaya, T.; Fujita, T.; Noiri, E. Urinary L-type fatty acid-binding protein can reflect renal tubulointerstitial injury. Am. J. Pathol. 2009, 174, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Makhloufi, C.; Nicolas, F.; McKay, N.; Fernandez, S.; Hache, G.; Garrigue, P.; Brunet, P.; Guillet, B.; Burtey, S.; Poitevin, S. Female AhR Knockout Mice Develop a Minor Renal Insufficiency in an Adenine-Diet Model of Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toda, T.; Saito, N.; Ikarashi, N.; Ito, K.; Yamamoto, M.; Ishige, A.; Watanabe, K.; Sugiyama, K. Intestinal flora induces the expression of Cyp3a in the mouse liver. Xenobiotica 2009, 39, 323–334. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W., Jr.; Kamanaka, M.; Booth, C.J.; Town, T.; Nakae, S.; Iwakura, Y.; Kolls, J.K.; Flavell, R.A. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 2009, 10, 603–609. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Koga, T.; Ichinose, K.; Tsokos, G.C. T cells and IL-17 in lupus nephritis. Clin. Immunol. 2017, 185, 95–99. [Google Scholar] [CrossRef]
- Ramani, K.; Tan, R.J.; Zhou, D.; Coleman, B.M.; Jawale, C.V.; Liu, Y.; Biswas, P.S. IL-17 Receptor Signaling Negatively Regulates the Development of Tubulointerstitial Fibrosis in the Kidney. Mediat. Inflamm. 2018, 2018, 5103672. [Google Scholar] [CrossRef] [Green Version]
- Rosendahl, A.; Kabiri, R.; Bode, M.; Cai, A.; Klinge, S.; Ehmke, H.; Mittrucker, H.W.; Wenzel, U.O. Adaptive immunity and IL-17A are not involved in the progression of chronic kidney disease after 5/6 nephrectomy in mice. Br. J. Pharmacol. 2019, 176, 2002–2014. [Google Scholar] [CrossRef]
- Al-Asmakh, M.; Zadjali, F. Use of Germ-Free Animal Models in Microbiota-Related Research. J. Microbiol. Biotechnol. 2015, 25, 1583–1588. [Google Scholar] [CrossRef] [Green Version]
- Haugen, A.C.; Schug, T.T.; Collman, G.; Heindel, J.J. Evolution of DOHaD: The impact of environmental health sciences. J. Dev. Orig. Health Dis. 2015, 6, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, T.; Sun, S.L.; Fujita, T.; Yamaki, S.; Asao, A.; Takahashi, T.; So, T.; Ishii, N. Homeostatic proliferation of naive CD4+ T cells in mesenteric lymph nodes generates gut-tropic Th17 cells. J. Immunol. 2013, 190, 5788–5798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, S.; Komiyama, Y.; Nambu, A.; Sudo, K.; Iwase, M.; Homma, I.; Sekikawa, K.; Asano, M.; Iwakura, Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 2002, 17, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Yokozawa, T.; Oura, H.; Zheng, P.D.; Fukase, M.; Koizumi, F.; Kanaoka, M. Metabolic Effects of Dietary Purine and Pyrimidine Bases in Rats. Agric. Biol. Chem. 1983, 47, 1297–1304. [Google Scholar] [CrossRef]
- Hill, D.A.; Siracusa, M.C.; Abt, M.C.; Kim, B.S.; Kobuley, D.; Kubo, M.; Kambayashi, T.; Larosa, D.F.; Renner, E.D.; Orange, J.S.; et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat. Med. 2012, 18, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Mishima, E.; Jinno, D.; Akiyama, Y.; Itoh, K.; Nankumo, S.; Shima, H.; Kikuchi, K.; Takeuchi, Y.; Elkordy, A.; Suzuki, T.; et al. Immuno-Northern Blotting: Detection of RNA Modifications by Using Antibodies against Modified Nucleosides. PLoS ONE 2015, 10, e0143756. [Google Scholar] [CrossRef] [Green Version]
- Mishima, E.; Sato, E.; Ito, J.; Yamada, K.I.; Suzuki, C.; Oikawa, Y.; Matsuhashi, T.; Kikuchi, K.; Toyohara, T.; Suzuki, T.; et al. Drugs Repurposed as Antiferroptosis Agents Suppress Organ Damage, Including AKI, by Functioning as Lipid Peroxyl Radical Scavengers. J. Am. Soc. Nephrol. 2020, 31, 280–296. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishima, E.; Ichijo, M.; Kawabe, T.; Kikuchi, K.; Akiyama, Y.; Toyohara, T.; Suzuki, T.; Suzuki, C.; Asao, A.; Ishii, N.; et al. Germ-Free Conditions Modulate Host Purine Metabolism, Exacerbating Adenine-Induced Kidney Damage. Toxins 2020, 12, 547. https://doi.org/10.3390/toxins12090547
Mishima E, Ichijo M, Kawabe T, Kikuchi K, Akiyama Y, Toyohara T, Suzuki T, Suzuki C, Asao A, Ishii N, et al. Germ-Free Conditions Modulate Host Purine Metabolism, Exacerbating Adenine-Induced Kidney Damage. Toxins. 2020; 12(9):547. https://doi.org/10.3390/toxins12090547
Chicago/Turabian StyleMishima, Eikan, Mariko Ichijo, Takeshi Kawabe, Koichi Kikuchi, Yukako Akiyama, Takafumi Toyohara, Takehiro Suzuki, Chitose Suzuki, Atsuko Asao, Naoto Ishii, and et al. 2020. "Germ-Free Conditions Modulate Host Purine Metabolism, Exacerbating Adenine-Induced Kidney Damage" Toxins 12, no. 9: 547. https://doi.org/10.3390/toxins12090547
APA StyleMishima, E., Ichijo, M., Kawabe, T., Kikuchi, K., Akiyama, Y., Toyohara, T., Suzuki, T., Suzuki, C., Asao, A., Ishii, N., Fukuda, S., & Abe, T. (2020). Germ-Free Conditions Modulate Host Purine Metabolism, Exacerbating Adenine-Induced Kidney Damage. Toxins, 12(9), 547. https://doi.org/10.3390/toxins12090547