The Anemonia sulcata Toxin BDS-I Protects Astrocytes Exposed to Aβ1–42 Oligomers by Restoring [Ca2+]i Transients and ER Ca2+ Signaling


 ,
, 
Abstract
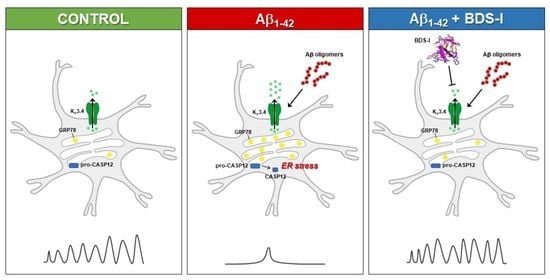
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
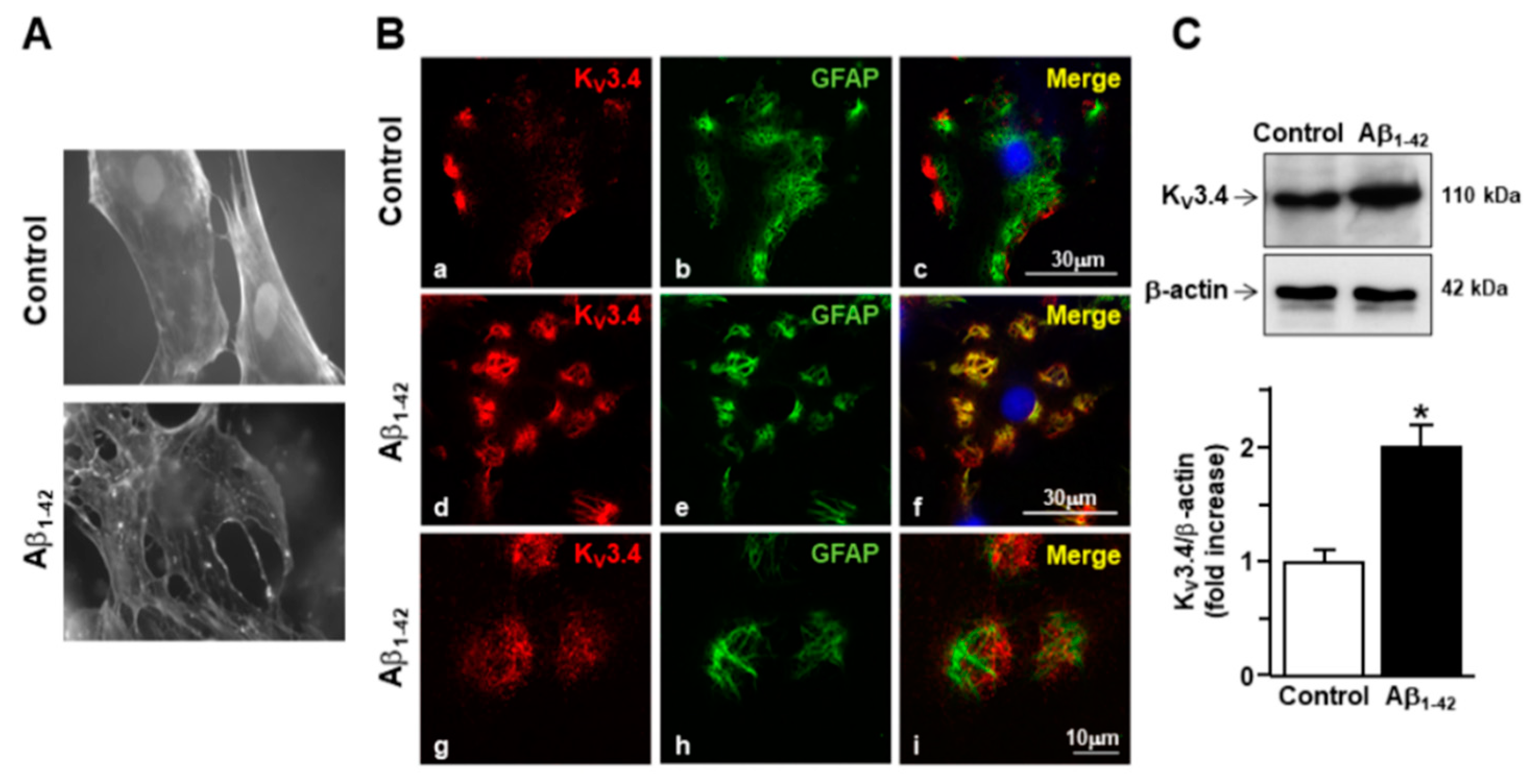
2.1. Exposure to Aβ1–42 Oligomers Upregulated KV3.4 Protein Expression and Activity in Activated Rat Primary Cortical Astrocytes
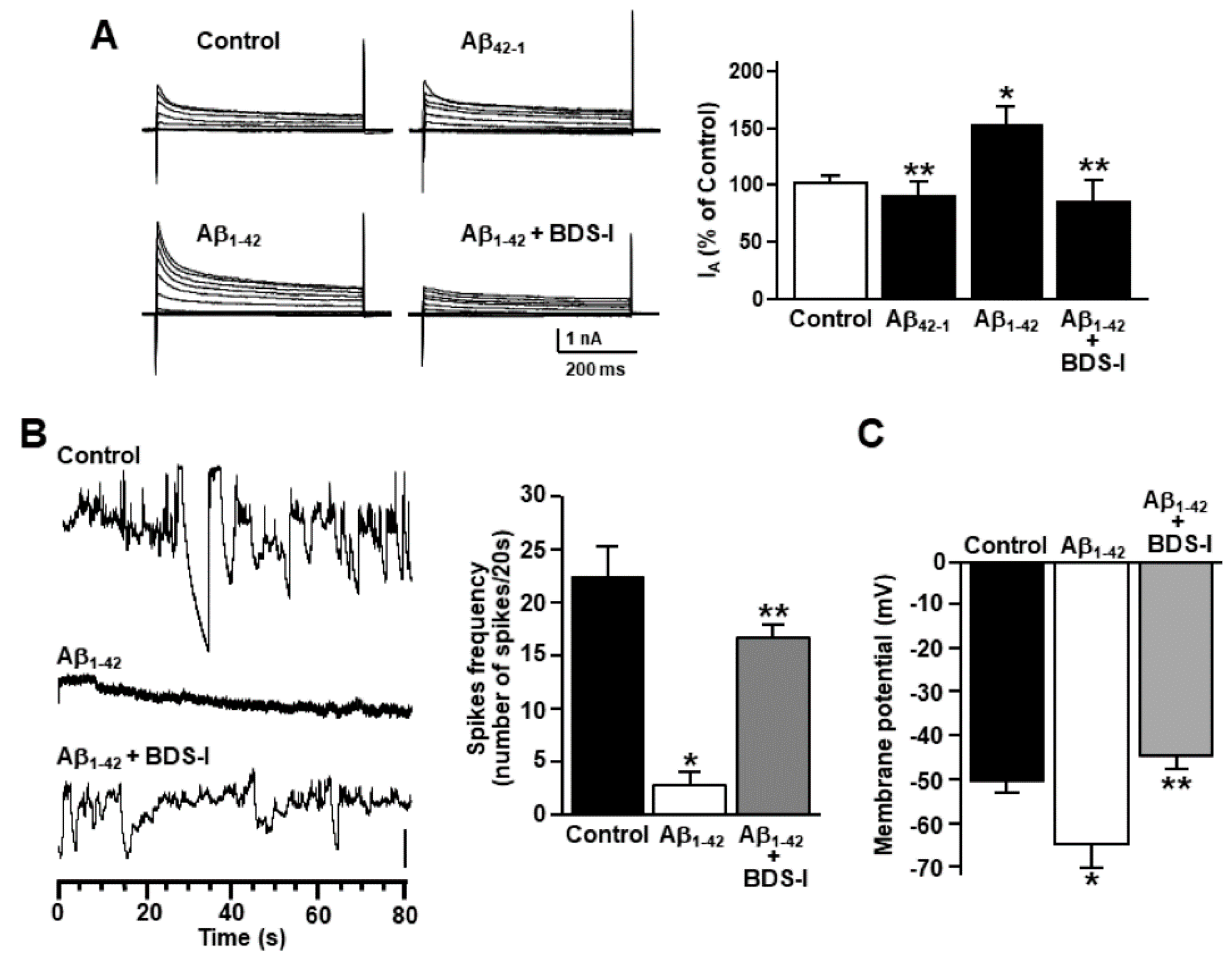
2.2. Effects of BDS-I on [Ca2+]i Transients and ER Ca2+Signaling in Rat Primary Astrocytes Exposed to Aβ1–42 Oligomers
2.3. Effects of BDS-I on ER Stress Markers in Rat Primary Astrocytes Exposed to Aβ1–42 Oligomers
2.4. Effects of BDS-I on ROS Production, Mitochondrial Activity, and Lactate Dehydrogenase (LDH) Release in Rat Primary Astrocytes Exposed to Aβ1–42 Oligomers
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Rat Primary Astrocytes
4.3. Solubilization of Aβ1–42 Peptide and Cellular Treatment
4.4. Western Blotting
4.5. Immunohistochemistry
4.6. Assessment of Nuclear and Cytoskeletal Morphology
4.7. Electrophysiology
4.8. [Ca2+]i Measurement
4.9. Measurement of Reactive Oxygen Species
4.10. Determination of Mitochondrial Activity
4.11. LDH Release Assay
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 22, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziato, L.; Boscia, F.; Pignataro, G. Ionic transporter activity in astrocytes, microglia, and oligodendrocytes during brain ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 969–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscia, F.; Begum, G.; Pignataro, G.; Sirabella, R.; Cuomo, O.; Casamassa, A.; Sun, D.; Annunziato, L. Glial Na+-dependent ion transporters in pathophysiological conditions. Glia 2016, 64, 1677–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Butt, A. Glial Physiology and Pathophysiology; Wiley–Blackwell: Oxford, UK, 2013. [Google Scholar]
- Pongs, O. Voltage-gated potassium channels: From hyperexcitability to excitement. FEBS Lett. 1999, 452, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Beiersdorfer, A.; Scheller, A.; Kirchhoff, F.; Lohr, C. Panglial gap junction between astrocytes and olfactory ensheathing cells mediate transmission of Ca2+ transients and neurovascular coupling. Glia 2019, 67, 1385–1400. [Google Scholar] [CrossRef]
- John, G.R.; Scemes, E.; Suadicani, S.O.; Liu, J.S.; Charles, P.C.; Lee, S.C.; Spray, D.C.; Brosnan, C.F. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc. Natl. Acad. Sci. USA 1999, 96, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Dani, J.W.; Chernjavsky, A.; Smith, S.J. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 1992, 8, 429–440. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Volterra, A.; Liaudet, N.; Savtchouk, I. Astrocyte Ca2+ signaling: An unexpected complexity. Nat. Rev. Neurosci. 2014, 15, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panatier, A.; Robitaille, R. Astrocytic mGluR5 and the tripartite synapse. Neuroscience 2016, 323, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; McCarthy, K.D. Astrocyte calcium signaling from observations to functions and the challenges therein. Cold Spring Harb. Perspect. Biol. 2015, 7, a020404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusakov, D.A. Disentangling calcium-driven astrocyte physiology. Nat. Rev. Neurosci. 2015, 16, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Patel, S.; Khakh, B.S. Probing the complexities of astrocyte calcium signaling. Trends Cell Biol. 2016, 26, 300–312. [Google Scholar] [CrossRef] [Green Version]
- Semyanov, A. Spatiotemporal pattern of calcium activity in astrocytic network. Cell Calcium 2019, 78, 15–25. [Google Scholar] [CrossRef]
- Hertz, L. Possible role of neuroglia: A potassium-mediated neuronal-neuroglial-neuronal impulse transmission system. Nature 1965, 206, 1091–1094. [Google Scholar] [CrossRef]
- Orkland, R.K.; Nicolls, G.J.; Kuffler, S.W. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966, 29, 788–806. [Google Scholar] [CrossRef]
- David, Y.; Cacheaux, L.P.; Ivens, S.; Lapilover, E.; Heinemann, U.; Kaufer, D.; Friedman, A. Astrocytic dysfunction in epileptogenesis: Consequence of altered potassium and glutamate homeostasis? J. Neurosci. 2009, 29, 10588–10599. [Google Scholar] [CrossRef] [Green Version]
- Leis, J.A.; Bekar, L.K.; Walz, W. Potassium homeostasis in the ischemic brain. Glia 2005, 50, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M.V.; Cavaliere, F.; Rodríguez, J.J.; Verkhratsky, A.; Matute, C. Ca2+-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A. Astroglial Calcium Signaling in Aging and Alzheimer’s Disease. Cold Spring Harb. Perspect. Biol. 2019, 11, a035188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Abeta production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s disease. Neurotherapeutics 2010, 7, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef]
- Boscia, F.; Pannaccione, A.; Ciccone, R.; Casamassa, A.; Franco, C.; Piccialli, I.; de Rosa, V.; Vinciguerra, A.; Di Renzo, G.; Annunziato, L. The expression and activity of KV3.4 channel subunits are precociously upregulated in astrocytes exposed to Aβ oligomers and in astrocytes of Alzheimer’s disease Tg2576 mice. Neurobiol. Aging 2017, 54, 187–198. [Google Scholar] [CrossRef]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Ojala, J.; Suuronen, T.; Kaarniranta, K.; Kauppinen, A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J. Cell. Mol. Med. 2008, 12, 2255–2262. [Google Scholar] [CrossRef]
- Ebrahimi, T.; Rust, M.; Kaiser, S.N.; Slowik, A.; Beyer, C.; Koczulla, A.R.; Schulz, J.B.; Habib, P.; Bach, J.P. α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1–42-stimulated murine astrocytes. J. Neuroinflamm. 2018, 15, 282. [Google Scholar] [CrossRef] [PubMed]
- Angulo, E.; Noé, V.; Casadó, V.; Mallol, J.; Gomez-Isla, T.; Lluis, C.; Ferrer, I.; Ciudad, C.J.; Franco, R. Up-regulation of the Kv3.4 potassium channel subunit in early stages of Alzheimer’s disease. J. Neurochem. 2004, 91, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Pannaccione, A.; Secondo, A.; Scorziello, A.; Calì, G.; Taglialatela, M.; Annunziato, L. Nuclear factor-kappaB activation by reactive oxygen species mediates voltage-gated K+ current enhancement by neurotoxic beta-amyloid peptides in nerve growth factor-differentiated PC-12 cells and hippocampal neurones. J. Neurochem. 2005, 94, 572–586. [Google Scholar] [CrossRef]
- Pannaccione, A.; Boscia, F.; Scorziello, A.; Adornetto, A.; Castaldo, P.; Sirabella, R.; Taglialatela, M.; Di Renzo, G.F.; Annunziato, L. Up-regulation and increased activity of KV3.4 channels and their accessory subunit MinK-related peptide 2 induced by amyloid peptide are involved in apoptotic neuronal death. Mol. Pharmacol. 2007, 72, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.P. Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 2003, 70, 363–386. [Google Scholar] [CrossRef]
- Diochot, S.; Schweitz, H.; Béress, L.; Lazdunski, M. Sea anemone peptides with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J. Biol. Chem. 1998, 273, 6744–6749. [Google Scholar] [CrossRef] [Green Version]
- Ciccone, R.; Piccialli, I.; Grieco, P.; Merlino, F.; Annunziato, L.; Pannaccione, A. Synthesis and pharmacological evaluation of a novel peptide based on Anemonia sulcata BDS-I Toxin as a new KV3.4 inhibitor exerting a neuroprotective effect against amyloid-β Peptide. Front. Chem. 2019, 7, 479. [Google Scholar] [CrossRef]
- Finol-Urdaneta, R.K.; Belovanovic, A.; Micic-Vicovac, M.; Kinsella, G.K.; McArthur, J.R.; Al-SabiMarine, A. Toxins Targeting Kv1 Channels: Pharmacological Tools and Therapeutic Scaffolds. Mar. Drugs 2020, 18, 173. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, T.; Han, X.; Deane, R.; Zlokovic, B.; Nedergaard, M. Two–photon imaging of astrocytic Ca2+ signaling and microvasculature in experimental mice models of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2007, 1097, 40–50. [Google Scholar] [CrossRef]
- Liu, P.; Jo, S.; Bean, B.P. Modulation of neuronal sodium channels by the sea anemone peptide BDS-I. J. Neurophysiol. 2012, 107, 3155–3167. [Google Scholar] [CrossRef] [Green Version]
- Frelin, C.; Vigne, P.; Schweitz, H.; Lazdunski, M. The interaction of sea anemone and scorpion neurotoxins with tetrodotoxin-resistant Na+ channels in rat myoblasts. A comparison with Na+ channels in other excitable and non–excitable cells. Mol. Pharmacol. 1984, 26, 70–74. [Google Scholar]
- Norton, R.S. Structure and structure-function relationships of sea anemone proteins that interact with the sodium channel. Toxicon 1991, 29, 1051–1084. [Google Scholar] [CrossRef]
- Vincent, J.P.; Balerna, M.; Barhanin, J.; Fosset, M.; Lazdunski, M. Binding of sea anemone toxin to receptor sites associated with gating system of sodium channel in synaptic nerve endings in vitro. Proc. Natl. Acad. Sci. USA 1980, 77, 1646–1650. [Google Scholar] [CrossRef] [Green Version]
- Lim, D.; Rodriguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Zeidán-Chuliá, F.; Genazzani, A.A.; Verkhratsky, A. Calcium signalling toolkits in astrocytes and spatio-temporal progression of Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 359–369. [Google Scholar] [CrossRef]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid β deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Dmitry Lim, D. Differential deregulation of astrocytic calcium signaling by amyloid-β, TNFα, IL-1 β and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Trkov, S.; Lasic, E.; Terzieva, S.; Kreft, M.; Arellano, J.J.R.; Parpura, V.; Verkhratsky, A. Expression of familial Alzheimer disease presenilin 1 gene attenuates vesicle traffic and reduces peptide secretion in cultured astrocytes devoid of pathologic tissue environment. Glia 2016, 64, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, M.S.; Gorbatyuk, O.S. The Molecular Chaperone GRP78/BiP as a Therapeutic Target for Neurodegenerative Disorders: A Mini Review. J. Genet. Syndr. Gene Ther. 2013, 4, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, J.A.; Tiffany-Castiglioni, E. The chaperone Grp78 in protein folding disorders of the nervous system. Neurochem. Res. 2015, 40, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Secondo, A.; Taglialatela, M.; Cataldi, M.; Giorgio, G.; Valore, M.; Di Renzo, G.; Annunziato, L. Pharmacological blockade of ERG K+ channels and Ca2+ influx through store-operated channels exerts opposite effects on intracellular Ca2+ oscillations in pituitary GH3 cells. Mol. Pharmacol. 2000, 58, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Yu, P.; Lu, Q.; Geller, H.M.; Yu, Z.; Chen, H. KCa3.1 constitutes a pharmacological target for astrogliosis associated with Alzheimer’s disease. Mol. Cell. 2016, 76, 21–32. [Google Scholar] [CrossRef]
- Yu, Z.; Dou, F.; Wang, Y.; Hou, L.; Chen, H. Ca2+-dependent endoplasmic reticulum stress correlation with astrogliosis involves upregulation of KCa3.1 and inhibition of AKT/mTOR signaling. J. Neuroinflamm. 2018, 15, 316. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Rodriguez-Arellano, J.J.; Zorec, R. Astroglia in Alzheimer’s disease. Adv. Exp. Med. Biol. 2019, 1175, 237–324. [Google Scholar]
- Boscia, F.; Gala, R.; Pannaccione, A.; Secondo, A.; Scorziello, A.; Di Renzo, G.; Annunziato, L. NCX1 expression and functional activity increase in microglia invading the infarct core. Stroke 2009, 40, 3608–3617. [Google Scholar] [CrossRef] [Green Version]
- Boscia, F.; D’Avanzo, C.; Pannaccione, A.; Secondo, A.; Casamassa, A.; Formisano, L.; Guida, N.; Sokolow, S.; Herchuelz, A.; Annunziato, L. Silencing or knocking out the Na+/Ca2+ exchanger-3 (NCX3) impairs oligodendrocyte differentiation. Cell Death Differ. 2012, 19, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Florio, E.; Keller, S.; Coretti, L.; Affinito, O.; Scala, G.; Errico, F.; Fico, A.; Boscia, F.; Sisalli, M.J.; Reccia, M.G.; et al. Tracking the evolution of epialleles during neural differentiation and brain development: D-Aspartate oxidase as a model gene. Epigenetics 2017, 12, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, R.; Franco, C.; Piccialli, I.; Boscia, F.; Casamassa, A.; de Rosa, V.; Cepparulo, P.; Cataldi, M.; Annunziato, L.; Pannaccione, A. Amyloid β-Induced Upregulation of Nav1.6 Underlies Neuronal Hyperactivity in Tg2576 Alzheimer’s Disease Mouse Model. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Stine, W.B.; Dahlgren, K.D.; Krafft, G.A.; LaDu, M.J. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 2003, 278, 11612–11622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, F.; Boscia, F.; Gigantino, V.; Tornincasa, M.; Fusco, A.; Franco, R.; Chieffi, P. The high-mobility group A1-estrogen receptor β nuclear interaction is impaired in human testicular seminomas. J. Cell Physiol. 2012, 227, 3749–3755. [Google Scholar] [CrossRef]
- Boscia, F.; Ferraguti, F.; Moroni, F.; Annunziato, L.; Pellegrini-Giampietro, D.E. mGlu1α and CB1 receptors are co-expressed in a subset of interneurons in the CA1 region of organotypic hippocampal slice cultures and adult rat brain. Neuropharmacology 2008, 55, 428–439. [Google Scholar] [CrossRef]
- Boscia, F.; Esposito, C.L.; Casamassa, A.; de Franciscis, V.; Annunziato, L.; Cerchia, L. The isolectin IB4 binds RET receptor tyrosine kinase in microglia. J. Neurochem. 2013, 126, 428–436. [Google Scholar] [CrossRef]
- Levee, M.G.; Dabrowska, M.I.; Lelli, J.L.; Hinshaw, D.B. Actin polymerization and depolimerization during apoptosis in HL-60 cells. Am. J. Physiol. 1996, 271, 1981–1992. [Google Scholar] [CrossRef]
- Bedi, S.S.; Yang, Q.; Crook, R.J.; Du, J.; Wu, Z.; Fishman, H.M.; Grill, R.J.; Carlton, S.M.; Walters, E.T. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J. Neurosci. 2010, 30, 14870–14882. [Google Scholar] [CrossRef]
- Gunhanlar, N.; Shpak, G.; van der Kroeg, M.; Gouty-Colomer, L.A.; Munshi, S.T.; Lendemeijer, B.; Ghazvini, M.; Dupont, C.; Hoogendijk, W.J.G.; Gribnau, J.; et al. A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol. Psychiatry 2018, 23, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Secondo, A.; Pannaccione, A.; Cataldi, M.; Sirabella, R.; Formisano, L.; Di Renzo, G.; Annunziato, L. Nitric oxide induces [Ca2+]i oscillations in pituitary GH3 cells: Involvement of IDR and ERG K+ currents. Am. J. Physiol. Cell Physiol. 2006, 290, C233–C243. [Google Scholar] [CrossRef]
- Petrozziello, T.; Secondo, A.; Tedeschi, V.; Esposito, A.; Sisalli, M.; Scorziello, A.; Di Renzo, G.; Annunziato, L. ApoSOD1 lacking dismutase activity neuroprotects motor neurons exposed to beta-methylamino-L-alanine through the Ca2+/Akt/ERK1/2 prosurvival pathway. Cell Death Differ. 2017, 24, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.S.; Buckley, S.; Deloach, J.R. Detection of the lethal effects of T-2 mycotoxin on cells using a rapid colorimetric viability assay. Toxicol. Lett. 1987, 39, 301–312. [Google Scholar] [CrossRef]
- Amoroso, S.; Gioielli, A.; Cataldi, M.; Di Renzo, G.F.; Annunziato, L. In the neuronal cell line SH-SY5Y, oxidative stress-induced free radical overproduction causes cell death without any participation of intracellular Ca2+ increase. Biochim. Biophys. Acta 1999, 1452, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Yang, B.; Zhou, Y.; Yin, C.; Liu, T.; Qian, H.; Xing, G.; Wang, S.; Li, F.; Zhang, Y.; et al. Acute Methylmercury Exposure and the Hypoxia-Inducible Factor-1α Signaling Pathway under Normoxic Conditions in the Rat Brain and Astrocytes in vitro. Environ. Health Perspect. 2019, 127, 127006. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccialli, I.; Tedeschi, V.; Boscia, F.; Ciccone, R.; Casamassa, A.; de Rosa, V.; Grieco, P.; Secondo, A.; Pannaccione, A. The Anemonia sulcata Toxin BDS-I Protects Astrocytes Exposed to Aβ1–42 Oligomers by Restoring [Ca2+]i Transients and ER Ca2+ Signaling. Toxins 2021, 13, 20. https://doi.org/10.3390/toxins13010020
Piccialli I, Tedeschi V, Boscia F, Ciccone R, Casamassa A, de Rosa V, Grieco P, Secondo A, Pannaccione A. The Anemonia sulcata Toxin BDS-I Protects Astrocytes Exposed to Aβ1–42 Oligomers by Restoring [Ca2+]i Transients and ER Ca2+ Signaling. Toxins. 2021; 13(1):20. https://doi.org/10.3390/toxins13010020
Chicago/Turabian StylePiccialli, Ilaria, Valentina Tedeschi, Francesca Boscia, Roselia Ciccone, Antonella Casamassa, Valeria de Rosa, Paolo Grieco, Agnese Secondo, and Anna Pannaccione. 2021. "The Anemonia sulcata Toxin BDS-I Protects Astrocytes Exposed to Aβ1–42 Oligomers by Restoring [Ca2+]i Transients and ER Ca2+ Signaling" Toxins 13, no. 1: 20. https://doi.org/10.3390/toxins13010020
APA StylePiccialli, I., Tedeschi, V., Boscia, F., Ciccone, R., Casamassa, A., de Rosa, V., Grieco, P., Secondo, A., & Pannaccione, A. (2021). The Anemonia sulcata Toxin BDS-I Protects Astrocytes Exposed to Aβ1–42 Oligomers by Restoring [Ca2+]i Transients and ER Ca2+ Signaling. Toxins, 13(1), 20. https://doi.org/10.3390/toxins13010020