Transcriptomic Profile of the Cockle Cerastoderma edule Exposed to Seasonal Diarrhetic Shellfish Toxin Contamination

 ,
,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
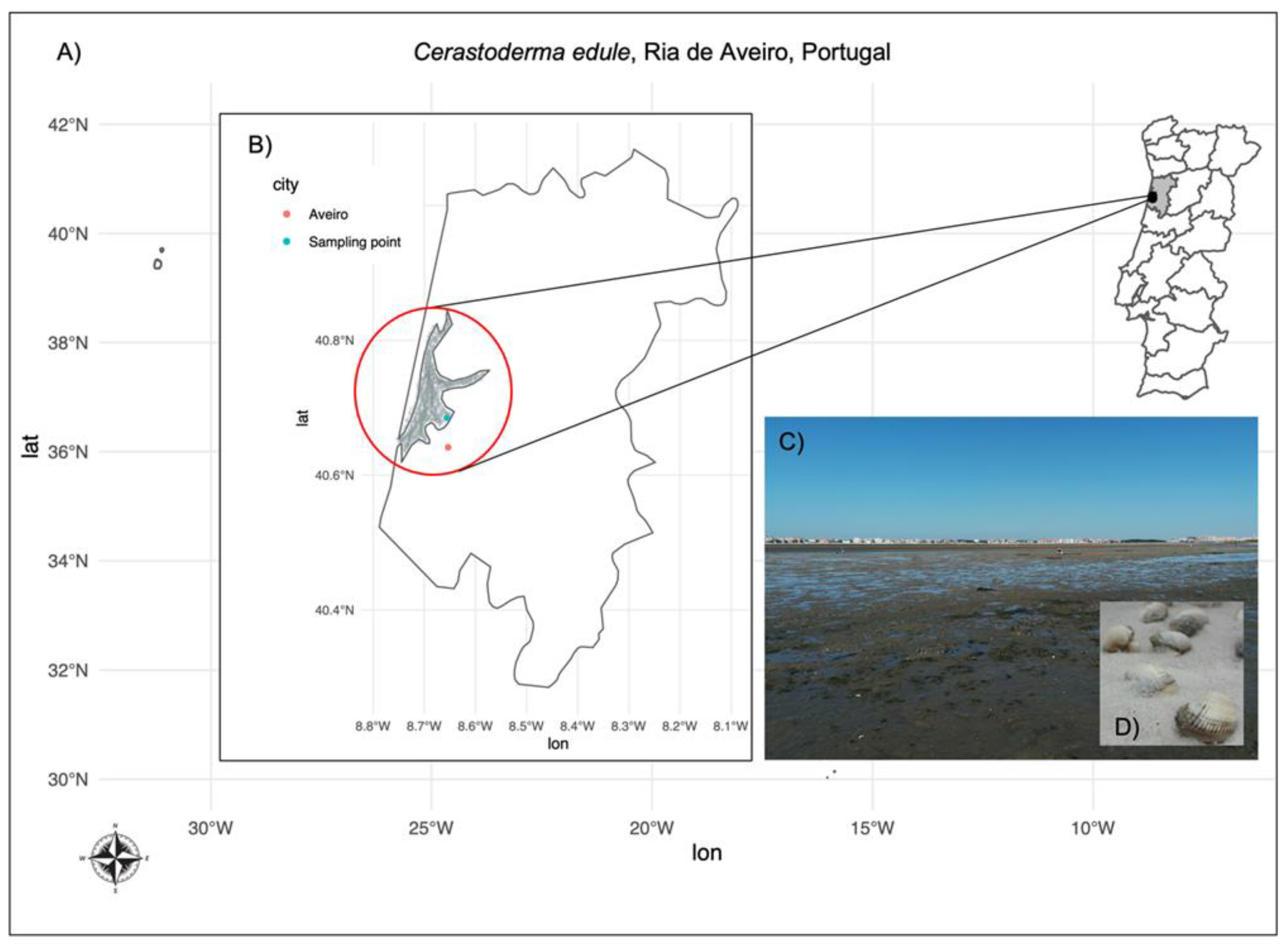
2.1. DSTs Determination by LC-MS/MS in C. edule Sampled in Ria de Aveiro, Portugal
2.2. Illumina Sequencing, Transcriptome Assembly and Completeness
2.3. Open Reading Frame Prediction and Annotation
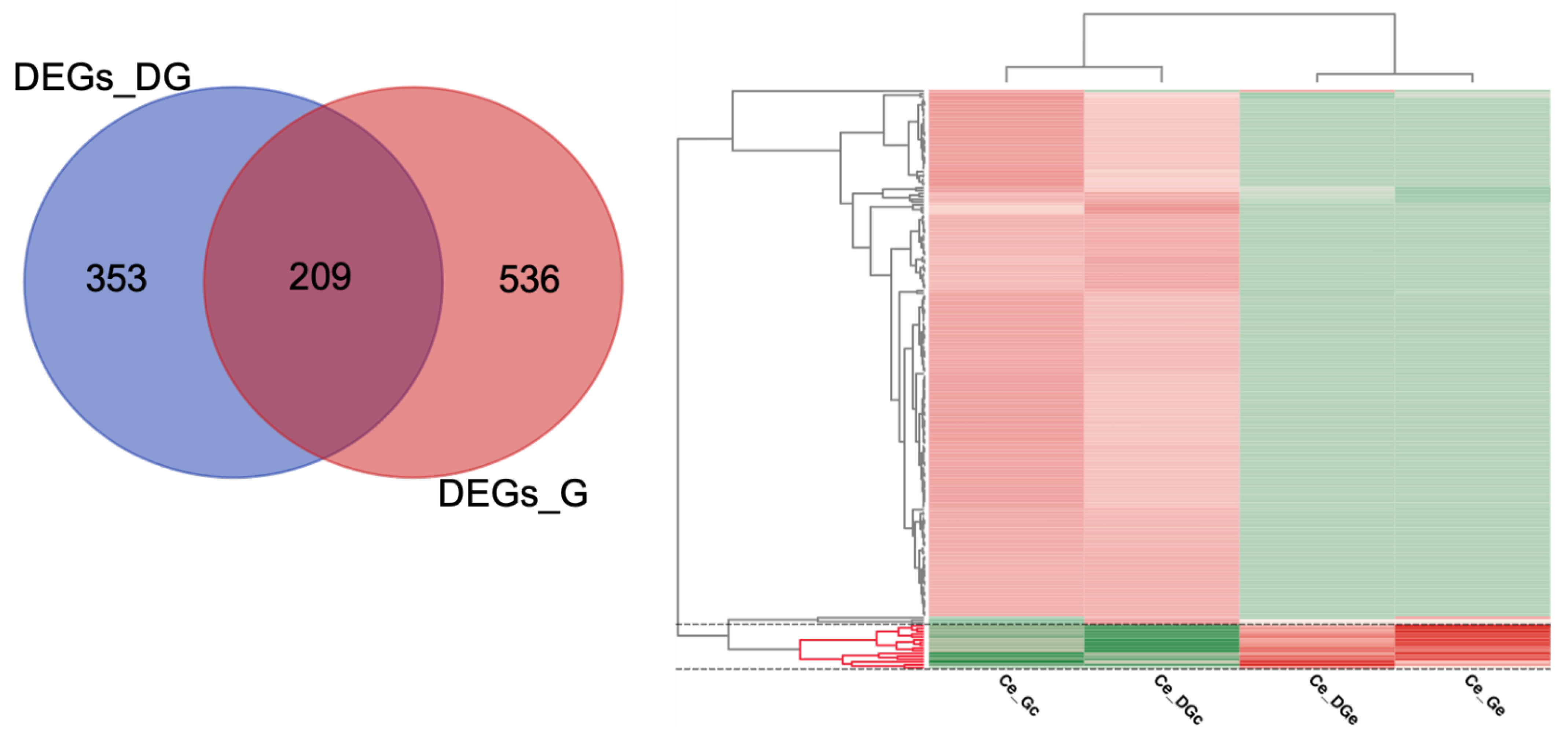
2.4. Differential Expression Analyses and Enrichment
3. Discussion
3.1. Common DEGs and Known Biomarkers Facing DSTs
3.2. Enriched Pathways and Degs Involved
4. Remarks
5. Materials and Methods
5.1. Sampling, RNA Extraction and RNASeq
5.2. DSTs Quantification by LC-MS/MS Analyses
5.3. RNA Extraction and Illumina Sequencing
5.4. Transcriptome Assembly and Completeness Assessment
5.5. Open Reading Frame Prediction and Transcript Abundance Estimation
5.6. Functional Annotation and Enrichment
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, D.M.; Fensin, E.; Gobler, C.J.; Hoeglund, A.E.; Hubbard, K.A.; Kulis, D.M.; Landsberg, J.H.; Lefebvre, K.A.; Provoost, P.; Richlen, M.L. Marine harmful algal blooms (HABs) in the United States: History, current status and future trends. Harmful Algae 2021, 102, 101975. [Google Scholar] [CrossRef] [PubMed]
- Berdalet, E.; Fleming, L.E.; Gowen, R.; Davidson, K.; Hess, P.; Backer, L.C.; Moore, S.K.; Hoagland, P.; Enevoldsen, H. Marine harmful algal blooms, human health and wellbeing: Challenges and opportunities in the 21st century. J. Mar. Biol. Assoc. UK 2016, 96, 61–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnan, E.; Arévalo, F.; Belin, C.; Branco, M.A.; Cembella, A.D.; Clarke, D.; Correa, J.; Davidson, K.; Dhanji-Rapkova, M.; Lozano, R.F. Diversity and regional distribution of harmful algal events along the Atlantic margin of Europe. Harmful Algae 2021, 102, 101976. [Google Scholar] [CrossRef] [PubMed]
- Lewitus, A.J.; Horner, R.A.; Caron, D.A.; Garcia-Mendoza, E.; Hickey, B.M.; Hunter, M.; Huppert, D.D.; Kudela, R.M.; Langlois, G.W.; Largier, J.L. Harmful algal blooms along the North American west coast region: History, trends, causes, and impacts. Harmful Algae 2012, 19, 133–159. [Google Scholar] [CrossRef] [Green Version]
- Yñiguez, A.T.; Lim, P.T.; Leaw, C.P.; Jipanin, S.J.; Iwataki, M.; Benico, G.; Azanza, R.V. Over 30 years of HABs in the Philippines and Malaysia: What have we learned? Harmful Algae 2021, 102, 101776. [Google Scholar] [CrossRef]
- Li, X.-Y.; Yu, R.-C.; Geng, H.-X.; Li, Y.-F. Increasing dominance of dinoflagellate red tides in the coastal waters of Yellow Sea, China. Mar. Pollut. Bull. 2021, 168, 112439. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, P.; Anderson, D.M.; Kaoru, Y.; White, A.W. The economic effects of harmful algal blooms in the United States: Estimates, assessment issues, and information needs. Estuaries 2002, 25, 819–837. [Google Scholar] [CrossRef]
- Van Dolah, F.M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Perspect. 2000, 108, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Reguera, B.; Riobó, P.; Rodríguez, F.; Díaz, P.A.; Pizarro, G.; Paz, B.; Franco, J.M.; Blanco, J. Dinophysis toxins: Causative organisms, distribution and fate in shellfish. Marine Drugs 2014, 12, 394–461. [Google Scholar] [CrossRef]
- Vale, P.; Botelho, M.J.; Rodrigues, S.M.; Gomes, S.S.; Sampayo, M.A.d.M. Two decades of marine biotoxin monitoring in bivalves from Portugal (1986–2006): A review of exposure assessment. Harmful Algae 2008, 7, 11–25. [Google Scholar]
- Bauder, A.G.; Cembella, A.D.; Bricelj, V.M.; Quilliam, M.A. Uptake and fate of diarrhetic shellfish poisoning toxins from the dinoflagellate Prorocentrum lima in the bay scallop Argopecten irradians. Marine Ecol. Prog. Ser. 2001, 213, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Shumway, S.E. Phycotoxin-related shellfish poisoning: Bivalve molluscs are not the only vectors. Rev. Fish. Sci. 1995, 3, 1–31. [Google Scholar] [CrossRef]
- Campos, A.; Freitas, M.; de Almeida, A.M.; Martins, J.C.; Domínguez-Pérez, D.; Osório, H.; Vasconcelos, V.; Reis Costa, P. OMICs approaches in diarrhetic shellfish toxins research. Toxins 2020, 12, 493. [Google Scholar] [CrossRef] [PubMed]
- García, C.; Schonstedt, V.; Santelices, J.P.; Lagos, N. High amount of dinophysistoxin-3 in Mytilus chilensis collected in Seno de Reloncaví, Chile, during massive human intoxication associated with outbreak of Vibrio parahaemolyticus. J. Toxicol. Sci. 2006, 31, 305. [Google Scholar] [PubMed] [Green Version]
- Yasumoto, T.; Murata, M.; Oshima, Y.; Sano, M.; Matsumoto, G.; Clardy, J. Diarrhetic shellfish toxins. Tetrahedron 1985, 41, 1019–1025. [Google Scholar] [CrossRef]
- Young, N.; Sharpe, R.A.; Barciela, R.; Nichols, G.; Davidson, K.; Berdalet, E.; Fleming, L.E. Marine harmful algal blooms and human health: A systematic scoping review. Harmful Algae 2020, 98, 101901. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, M.; Fujiki, H.; Okabe, S.; Nishiwaki, S.; Brautigan, D.; Ingebritsen, T.S.; Rosner, M.R. Structurally different members of the okadaic acid class selectively inhibit protein serine/threonine but not tyrosine phosphatase activity. Toxicon 1992, 30, 873–878. [Google Scholar] [CrossRef]
- Blanco, J.; Mariño, C.; Martín, H.; Acosta, C.P. Anatomical distribution of diarrhetic shellfish poisoning (DSP) toxins in the mussel Mytilus galloprovincialis. Toxicon 2007, 50, 1011–1018. [Google Scholar]
- McCarron, P.; Kilcoyne, J.; Hess, P. Effects of cooking and heat treatment on concentration and tissue distribution of okadaic acid and dinophysistoxin-2 in mussels (Mytilus edulis). Toxicon 2008, 51, 1081–1089. [Google Scholar]
- Prego-Faraldo, M.; Vieira, L.; Eirin-Lopez, J.; Méndez, J.; Guilhermino, L. Transcriptional and biochemical analysis of antioxidant enzymes in the mussel Mytilus galloprovincialis during experimental exposures to the toxic dinoflagellate Prorocentrum lima. Mar. Environ. Res. 2017, 129, 304–315. [Google Scholar]
- Wei, X.-M.; Lu, M.-Y.; Duan, G.-F.; Li, H.-Y.; Liu, J.-S.; Yang, W.-D. Responses of CYP450 in the mussel Perna viridis after short-term exposure to the DSP toxins-producing dinoflagellate Prorocentrum lima. Ecotoxicol. Environ. Saf. 2019, 176, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wei, X.-M.; Weng, H.-W.; Li, H.-Y.; Liu, J.-S.; Yang, W.-D. Expression profile of eight glutathione S-transferase genes in Crassostrea ariakensis after exposure to DSP toxins producing dinoflagellate Prorocentrum lima. Toxicon 2015, 105, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, S.-L.; Zheng, J.-W.; Li, H.-Y.; Liu, J.-S.; Yang, W.-D. P-glycoprotein and its inducible expression in three bivalve species after exposure to Prorocentrum lima. Aquat. Toxicol. 2015, 169, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, J.; Chen, W.-C.; Li, H.-Y.; Liu, J.-S.; Jiang, T.; Yang, W.-D. P-glycoprotein expression in Perna viridis after exposure to Prorocentrum lima, a dinoflagellate producing DSP toxins. Fish Shellfish Immunol. 2014, 39, 254–262. [Google Scholar] [CrossRef]
- Martínez-Escauriaza, R.; Lozano, V.; Pérez-Parallé, M.L.; Blanco, J.; Sánchez, J.L.; Pazos, A.J. Expression Analyses of Genes Related to Multixenobiotic Resistance in Mytilus galloprovincialis after Exposure to Okadaic Acid-Producing Dinophysis acuminata. Toxins 2021, 13, 614. [Google Scholar]
- Martins, J.C.; Domínguez-Pérez, D.; Azevedo, C.; Braga, A.C.; Costa, P.R.; Osório, H.; Vasconcelos, V.; Campos, A. Molecular Responses of Mussel Mytilus galloprovincialis Associated to Accumulation and Depuration of Marine Biotoxins Okadaic Acid and Dinophysistoxin-1 Revealed by Shotgun Proteomics. Front. Mar. Sci. 2020, 7, 589822. [Google Scholar] [CrossRef]
- Miao, J.; Chi, L.; Pan, L.; Song, Y. Generally detected genes in comparative transcriptomics in bivalves: Toward the identification of molecular markers of cellular stress response. Environ. Toxicol. Pharmacol. 2015, 39, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Ulloa, V.; Fernandez-Tajes, J.; Aguiar-Pulido, V.; Prego-Faraldo, M.V.; Florez-Barros, F.; Sexto-Iglesias, A.; Mendez, J.; Eirin-Lopez, J.M. Unbiased high-throughput characterization of mussel transcriptomic responses to sublethal concentrations of the biotoxin okadaic acid. PeerJ 2015, 3, e1429. [Google Scholar] [CrossRef] [Green Version]
- Murgarella, M.; Puiu, D.; Novoa, B.; Figueras, A.; Posada, D.; Canchaya, C. A first insight into the genome of the filter-feeder mussel Mytilus galloprovincialis. PLoS ONE 2016, 11, e0151561. [Google Scholar]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.-L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Sellami, B.; Khazri, A.; Louati, H.; Dellali, M.; Driss, M.R.; Aïssa, P.; Mahmoudi, E.; Hamouda, B.; Coelho, A.V.; Sheehan, D. Effects of anthracene on filtration rates, antioxidant defense system, and redox proteomics in the Mediterranean clam Ruditapes decussatus (Mollusca: Bivalvia). Environ. Sci. Pollut. Res. 2015, 22, 10956–10968. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Pérez, D.; Lippolis, J.; Dennis, M.; Miller, B.; Tiley, K.; Vasconcelos, V.; de Almeida, A.M.; Campos, A. The Queen Conch (Lobatus gigas) proteome: A valuable tool for biological studies in marine gastropods. Protein J. 2019, 38, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Vale, P. Differential dynamics of dinophysistoxins and pectenotoxins between blue mussel and common cockle: A phenomenon originating from the complex toxin profile of Dinophysis acuta. Toxicon 2004, 44, 123–134. [Google Scholar] [CrossRef]
- Rossignoli, A.E.; Fernández, D.; Regueiro, J.; Mariño, C.; Blanco, J. Esterification of okadaic acid in the mussel Mytilus galloprovincialis. Toxicon 2011, 57, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Vale, P.; Sampayo, M.A.d.M. First confirmation of human diarrhoeic poisonings by okadaic acid esters after ingestion of razor clams (Solen marginatus) and green crabs (Carcinus maenas) in Aveiro lagoon, Portugal and detection of okadaic acid esters in phytoplankton. Toxicon 2002, 40, 989–996. [Google Scholar] [CrossRef]
- Suzuki, T.; Mitsuya, T. Comparison of dinophysistoxin-1 and esterified dinophysistoxin-1 (dinophysistoxin-3) contents in the scallop Patinopecten yessoensis and the mussel Mytilus galloprovincialis. Toxicon 2001, 39, 905–908. [Google Scholar] [CrossRef]
- Braga, A.C.; Lage, S.; Pacheco, M.; Rydberg, S.; Costa, P.R. Native (Ruditapes decussatus) and non-indigenous (R. philippinarum) shellfish species living in sympatry: Comparison of regulated and non-regulated biotoxins accumulation. Mar. Environ. Res. 2017, 129, 147–155. [Google Scholar]
- Vale, P.; Sampayo, M.A.d.M. Seasonality of diarrhetic shellfish poisoning at a coastal lagoon in Portugal: Rainfall patterns and folk wisdom. Toxicon 2003, 41, 187–197. [Google Scholar] [CrossRef]
- Oyaneder-Terrazas, J.; Polanco, C.; Figueroa, D.; Barriga, A.; García, C. In vitro biotransformation of OA-group and PTX-group toxins in visceral and non-visceral tissues of Mytilus chilensis and Ameghinomya antiqua. Food Addit. Contam. Part A 2020, 37, 1216–1228. [Google Scholar]
- The UniProt Consortium. The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar]
- BioBam Bioinformatics. OmicsBox-Bioinformatics Made Easy (Version 1.4.11). Available online: https://www.biobam.com/omicsbox (accessed on 3 March 2019).
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prego-Faraldo, M.V.; Martínez, L.; Méndez, J. RNA-Seq analysis for assessing the early response to DSP toxins in Mytilus galloprovincialis digestive gland and gill. Toxins 2018, 10, 417. [Google Scholar]
- Nielsen, P.; Krock, B.; Hansen, P.J.; Vismann, B. Effects of the DSP-toxic dinoflagellate Dinophysis acuta on clearance and respiration rate of the blue mussel, Mytilus edulis. PLoS ONE 2020, 15, e0230176. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Zou, Y.; Weng, H.-w.; Li, H.-Y.; Liu, J.-S.; Yang, W.-D. Proteomic profile in Perna viridis after exposed to Prorocentrum lima, a dinoflagellate producing DSP toxins. Environ. Pollut. 2015, 196, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Holm, L.; Sander, C. The immunoglobulin fold: Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Halaby, D.; Poupon, A.; Mornon, J.-P. The immunoglobulin fold family: Sequence analysis and 3D structure comparisons. Protein Eng. 1999, 12, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Potapov, V.; Sobolev, V.; Edelman, M.; Kister, A.; Gelfand, I. Protein—Protein recognition: Juxtaposition of domain and interface cores in immunoglobulins and other sandwich-like proteins. J. Mol. Biol. 2004, 342, 665–679. [Google Scholar]
- Teichmann, S.A.; Chothia, C. Immunoglobulin superfamily proteins in Caenorhabditis elegans. J. Mol. Biol. 2000, 296, 1367–1383. [Google Scholar] [CrossRef] [Green Version]
- Bodian, D.L.; Jones, E.Y.; Harlos, K.; Stuart, D.I.; Davis, S.J. Crystal structure of the extracellular region of the human cell adhesion molecule CD2 at 2.5 Å resolution. Structure 1994, 2, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.K.; Xue, H. Sequence profiles of immunoglobulin and immunoglobulin-like domains. J. Mol. Biol. 1997, 274, 530–545. [Google Scholar]
- Sun, S.-C.; Lindstrom, I.; Boman, H.G.; Faye, I.; Schmidt, O. Hemolin: An insect-immune protein belonging to the immunoglobulin superfamily. Science 1990, 250, 1729–1732. [Google Scholar] [CrossRef]
- Ladendorff, N.E.; Kanost, M.R. Bacteria-induced protein P4 (hemolin) from Manduca sexta: A member of the immunoglobulin superfamily which can inhibit hemocyte aggregation. Arch. Insect Biochem. Physiol. 1991, 18, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Faye, I.; Lindström-Dinnetz, I.; Sun, S.-C. Specific immune recognition of insect hemolin. Dev. Comp. Immunol. 1993, 17, 195–200. [Google Scholar] [CrossRef]
- Aathmanathan, V.S.; Jothi, N.; Prajapati, V.K.; Krishnan, M. Investigation of immunogenic properties of Hemolin from silkworm, Bombyx mori as carrier protein: An immunoinformatic approach. Sci. Rep. 2018, 8, 6957. [Google Scholar] [CrossRef]
- Bettencourt, R.; Lanz-Mendoza, H.; Lindquist, K.R.; Faye, I. Cell adhesion properties of hemolin, an insect immune protein in the Ig superfamily. Eur. J. Biochem. 1997, 250, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Watthanasurorot, A.; Jiravanichpaisal, P.; Liu, H.; Söderhäll, I.; Söderhäll, K. Bacteria-induced Dscam isoforms of the crustacean, Pacifastacus leniusculus. PLoS Pathog. 2011, 7, e1002062. [Google Scholar] [CrossRef]
- Hoek, R.M.; Smit, A.B.; Frings, H.; Vink, J.M.; de Jong-Brink, M.; Geraerts, W.P. A new Ig-superfamily member, molluscan defence molecule (MDM) from Lymnaea stagnalis, is down-regulated during parasitosis. Eur. J. Immunol. 1996, 26, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-M.; Kang, S.; Ahn, D.-H.; Kim, J.-H.; Ahn, I.; Lee, C.-W.; Cho, J.-L.; Min, G.-S.; Park, H. First insights into the subterranean crustacean Bathynellacea transcriptome: Transcriptionally reduced opsin repertoire and evidence of conserved homeostasis regulatory mechanisms. PLoS ONE 2017, 12, e0170424. [Google Scholar] [CrossRef]
- Magalhães, L.; Correia, S.; de Montaudouin, X.; Freitas, R. Spatio-temporal variation of trematode parasites community in Cerastoderma edule cockles from Ria de Aveiro (Portugal). Environ. Res. 2018, 164, 114–123. [Google Scholar] [CrossRef]
- De Sousa Magalhães, L.V. Inventory, Dynamics and Impact of the Trematodes Parasites in Bivalves with High Economic Importance. Ph.D. Thesis, University of Bordeaux, Bordeaux, France, 2018. [Google Scholar]
- Dou, M.; Jiao, Y.-h.; Zheng, J.-w.; Zhang, G.; Li, H.-y.; Liu, J.-s.; Yang, W.-d. De novo transcriptome analysis of the mussel Perna viridis after exposure to the toxic dinoflagellate Prorocentrum lima. Ecotoxicol. Environ. Saf. 2020, 192, 110265. [Google Scholar]
- Gerdol, M.; Venier, P.; Pallavicini, A. The genome of the Pacific oyster Crassostrea gigas brings new insights on the massive expansion of the C1q gene family in Bivalvia. Dev. Comp. Immunol. 2015, 49, 59–71. [Google Scholar] [CrossRef]
- Kishore, U.; Gaboriaud, C.; Waters, P.; Shrive, A.K.; Greenhough, T.J.; Reid, K.B.; Sim, R.B.; Arlaud, G.J. C1q and tumor necrosis factor superfamily: Modularity and versatility. Trends Immunol. 2004, 25, 551–561. [Google Scholar] [CrossRef]
- Gestal, C.; Pallavicini, A.; Venier, P.; Novoa, B.; Figueras, A. MgC1q, a novel C1q-domain-containing protein involved in the immune response of Mytilus galloprovincialis. Dev. Comp. Immunol. 2010, 34, 926–934. [Google Scholar] [CrossRef] [Green Version]
- He, Z.-b.; Duan, G.-f.; Liang, C.-y.; Li, H.-y.; Liu, J.-s.; Yang, W.-d. Up-regulation of Nrf2-dependent antioxidant defenses in Perna viridis after exposed to Prorocentrum lima. Fish Shellfish Immunol. 2019, 90, 173–179. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [PubMed] [Green Version]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockwood, B.L.; Sanders, J.G.; Somero, G.N. Transcriptomic responses to heat stress in invasive and native blue mussels (genus Mytilus): Molecular correlates of invasive success. J. Exp. Biol. 2010, 213, 3548–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockwood, B.L.; Somero, G.N. Transcriptomic responses to salinity stress in invasive and native blue mussels (genus Mytilus). Mol. Ecol. 2011, 20, 517–529. [Google Scholar] [CrossRef]
- European Union Reference Laboratory for Marine Biotoxins (EU-RL-MB). EU-Harmonised Standard Operating Procedure for Determination of Lipophilic Marine Biotoxins in Molluscs by LC-MS/MS; Spanish Ministry of Consumer Affairs: Madrid, Spain, 2015. [Google Scholar]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data [Online]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 January 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Martis, M.M.; Tarbiat, B.; Tydén, E.; Jansson, D.S.; Höglund, J. RNA-Seq de novo assembly and differential transcriptome analysis of the nematode Ascaridia galli in relation to in vivo exposure to flubendazole. PLoS ONE 2017, 12, e0185182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from rNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. In Gene Prediction; Humana: New York, NY, USA, 2019; pp. 227–245. [Google Scholar]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Haas, B.; Papanicolaou, A. TransDecoder 5.5.0 [Online]. 2015. Available online: https://github.com/TransDecoder/TransDecoder/wiki (accessed on 3 February 2021).
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat Meth. Nat. Res. 2012, 9, 357–359. [Google Scholar]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roninson, M.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Summary of Analyses per Sample/Condition 1 | Statistics | Ce_DGc | Ce_DGe | Ce_Gc | Ce_Ge |
|---|---|---|---|---|---|
| Raw Data Statistics | Total Bases 2 | 6,248,071,054 | 6,402,268,630 | 6,258,018,330 | 6,333,078,920 |
| Read Count 3 | 41,377,954 | 42,399,130 | 41,443,830 | 41,940,920 | |
| GC (%) 4 | 42.33 | 41.63 | 41.09 | 41.05 | |
| AT (%) 5 | 57.67 | 58.37 | 58.91 | 58.95 | |
| Q20 (%) 6 | 97.95 | 97.89 | 98.02 | 97.9 | |
| Q30 (%) 7 | 94.06 | 93.83 | 93.98 | 93.8 | |
| de novoTranscriptome Assembly (Trinity v2.10.0) | Assembled transcripts | 684,723 | |||
| Total genes | 361,460.0 | ||||
| Minimum/Maximum Contig length (bp: base pair) 8 | 200/42,164 bp | ||||
| Contig length (N50) 8 | 1094 bp | ||||
| Contig length (Average) 9 | 736.09 bp | ||||
| GC (%) | 37.39 | ||||
| Total assembled bases (all transcripts) | 504,019,879 | ||||
| Total assembled bases (longest isoform per gene) | 199,701,724 | ||||
| Percentage of all read aligned/not aligned 10 | 92.1%/7.8% | 92.4%/7.5% | 93.1%/6.8% | 93.3%/6.6% | |
| Assembly Completeness (BUSCO v5) 11 | number of BUSCOs/species | 954/65 | |||
| Complete Single-Copy | 282/29.56% | ||||
| Complete duplicated | 658/69% | ||||
| Fragmented | 8/0.84% | ||||
| Missing | 6/0.63% | ||||
| Assembly Clustering 12 (CD-HIT 4.8.1) and Completeness 11 | number of clusters at 90% identity | 402,961 | |||
| Complete Single-Copy | 771/80.82% | ||||
| Complete Duplicated | 170/17.82% | ||||
| Fragmented | 8/0.84% | ||||
| Missing | 5/0.52% | ||||
| ORFs prediction 13 (TransDecoder v5.5.0) | Transcripts with ORF (best isoform per gene) | 48,107 | |||
| ORFs Annotation | Blast Hits nr/NCBI (Metazoa/non-Metazoa) 14 | 37,184/362 | |||
| Blast Hits SwissProt (Metazoa/non-Metazoa) 15 | 26,968/22 | ||||
| Averall ORFs with Blast hits/no hits 16 | 37,262 (77.5%)/10,845 (22.5%) | ||||
| EggNOG 17 | 31,848 (66.2%) | ||||
| KEGG Pathways/KO 18 | 12,760 (25.9%)/20,621 (42.8%) | ||||
| IPS/GO annotation (%) 19 | 48,107 (100%)/19,398 (40.3%) | ||||
| Gene Ontology (ORFs with GO annotation) 20 | 21,799 (45.3%) | ||||
| Functional 1 Categories | Gene/Transcript 2 | Gene/Transcript Description 3 | Gene/Transcript 4 GO/IP Description | Organism 5 (Accession) | DEGs Subset 6 (UP-Regulated/Down-Regulated) LFC 7 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Unique (DG) 6.1 185 UR | Unique (DG) 6.2 168 DR | Common 16 UR 6.3 | Common 189 DR 6.4 | Unique (G) 6.5 80 UR | Unique (G) 6.6 456 DR | |||||
| Antioxidant system | TRINITY_DN158227_c0_g1 | Extracellular superoxide dismutase [Cu-Zn] | Superoxide metabolic process; metal ion binding | Fasciolopsis buski(KAA0197766.1) | −8.15 | |||||
| TRINITY_DN250845_c0_g1 | Extracellular superoxide dismutase [Cu-Zn] | Fasciolopsis buski(KAA0197766.1) | −8.15 | |||||||
| TRINITY_DN57639_c0_g2 | Superoxide dismutase [Cu-Zn] | Elysia chlorotica (RUS83892.1) | −9.14 | |||||||
| TRINITY_DN54684_c0_g1 | Glutathione peroxidase | Response to oxidative stress; glutathione peroxidase activity | Clonorchis sinensis(GAA50772.1) | −7.91 | ||||||
| TRINITY_DN1482_c1_g1 | Glutathione hydrolase 1 proenzyme-like | Glutathione catabolic process glutathione hydrolase activity | Pecten maximus(XP_033752752.1) | 2.84 | ||||||
| TRINITY_DN7416_c0_g1 | Glutathione S-transferase Mu 3 | Glutathione metabolic process; protein binding | Alligator mississippiensis(XP_006260118.1) | −8.29 | ||||||
| TRINITY_DN8990_c0_g1 | Putative glutathione-specific γ-glutamyl cyclotransferase 2 | Glutathione catabolic process; γ-glutamyl cyclotransferase activity | Octopus vulgaris(XP_029637948.1) | 3.46 | ||||||
| Metabolic detoxification | TRINITY_DN9032_c0_g3 | Cytochrome P450 2U1-like | Monooxygenase activity; iron ion binding; oxidoreductase activity Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen; heme binding | Mizuhopecten yessoensis(XP_021373153.1) | 3.66 | |||||
| TRINITY_DN2028_c0_g1 | Steroid 17-α-hydroxylase /17,20 lyase-like | Pecten maximus (XP_033763830.1) | 5.2 | |||||||
| 3.1 | ||||||||||
| TRINITY_DN12127_c0_g1 | ABC transporter G family member 21-like isoform X1 | ATPase-coupled transmembrane transporter activity | Pomacea canaliculate(XP_025089510.1) | 6.64 | ||||||
| TRINITY_DN86_c1_g1 | Phosphatidylcholine translocator ABCB4-like | ATP binding; xenobiotic transmembrane transporter activity | Mizuhopecten yessoensis(XP_021367043.1) | 3.18 | ||||||
| Transcription | TRINITY_DN70033_c0_g1 | KH domain protein | RNA processing; DNA binding; RNA binding | Opisthorchis felineus(TGZ67600.1) | −8.01 | |||||
| −9.24 | ||||||||||
| TRINITY_DN22122_c0_g1 | Cell division cycle and apoptosis regulator protein 1 | regulation of transcription, DNA-templated | Paragonimus westermani(KAA3674315.1) | −9.21 | ||||||
| −10.38 | ||||||||||
| TRINITY_DN54844_c0_g2 | CREB-binding protein | regulation of transcription, DNA-templated; protein binding | Schistosoma japonicum(TNN09875.1) | −7.39 | ||||||
| TRINITY_DN116751_c0_g1 | Transcription elongationregulator 1 | FF domain; protein-protein interaction | Paragonimus westermani(KAA3677206.1) | −8.26 | ||||||
| −8.56 | ||||||||||
| cytoskeleton, actin related proteins | TRINITY_DN9687_c0_g1 | Tubulin α chain testis- specific | structural constituent of cytoskeleton; GTP binding; microtubule | Fasciola hepatica(KAA3677206.1) | −8.65 | |||||
| −8.66 | ||||||||||
| TRINITY_DN9687_c1_g2 | Tubulin α-chain | Fasciola hepatica(THD25403.1) | −8.48 | |||||||
| TRINITY_DN181044_c0_g1 | Actin depolymerizing factor | actin filament depolymerization; actin binding | Opisthorchis viverrine(XP_009175913.1) | −7.5 | ||||||
| TRINITY_DN34498_c0_g1 | Actin-1 isoform X2 | actin family | Rattus norvegicus(XP_002726247.1) | −7.08 | ||||||
| TRINITY_DN613_c1_g1 | VChain V, beta-actin | ATP binding; cytoplasm; cytoskeleton | Sus scrofa(pdb|5NW4|) | −11.08 | ||||||
| −11.81 | ||||||||||
| TRINITY_DN6932_c0_g1 | Actin β/γ 1 | Actin family | Clonorchis sinensis(GAA57823.1) | −8.61 | ||||||
| TRINITY_DN7730_c0_g1 | Actin β/γ 1 | Opisthorchis felineus(TGZ74009.1) | −5.42 | |||||||
| −7.54 | ||||||||||
| Inmune response | TRINITY_DN10313_c0_g2 | Complement C1Q-like protein | Protein binding | Sinonovacula constricta(QCO31675.1) | −3.83 | |||||
| TRINITY_DN1050_c0_g1 | Complement C1q tumor necrosis factor-related protein 4-like | Octopus vulgaris(XP_029647271.1) | 8.06 | |||||||
| TRINITY_DN14010_c0_g1 | Putative C1q domain containing protein MgC1q83 | Mytilus galloprovincialis(CBX41732.1) | −8.38 | |||||||
| TRINITY_DN1909_c0_g2 | Complement C1q-like protein 2 | Lates calcarifer (XP_018543622.1) | 3.91 | |||||||
| TRINITY_DN45102_c0_g1 | Complement C1Q-like protein | Pecten maximus(XP_033749093.1) | −5.41 | |||||||
| Heat shock protein HSP 90 (HSP90) | TRINITY_DN16801_c0_g1 | Hsp90 protein | Protein folding; ATP binding; ATP hydrolysis activity; unfolded protein binding | Opisthorchis viverrine(XP_009172981.1) | −8.31 | |||||
| −10.24 | ||||||||||
| TRINITY_DN260239_c0_g1 | Hsp90 co-chaperone Cdc37 isoform 1 | Protein kinase binding | Schistosoma japonicum(AAP06244.1) | −7.99 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez-Pérez, D.; Martins, J.C.; Almeida, D.; Costa, P.R.; Vasconcelos, V.; Campos, A. Transcriptomic Profile of the Cockle Cerastoderma edule Exposed to Seasonal Diarrhetic Shellfish Toxin Contamination. Toxins 2021, 13, 784. https://doi.org/10.3390/toxins13110784
Domínguez-Pérez D, Martins JC, Almeida D, Costa PR, Vasconcelos V, Campos A. Transcriptomic Profile of the Cockle Cerastoderma edule Exposed to Seasonal Diarrhetic Shellfish Toxin Contamination. Toxins. 2021; 13(11):784. https://doi.org/10.3390/toxins13110784
Chicago/Turabian StyleDomínguez-Pérez, Dany, José Carlos Martins, Daniela Almeida, Pedro Reis Costa, Vitor Vasconcelos, and Alexandre Campos. 2021. "Transcriptomic Profile of the Cockle Cerastoderma edule Exposed to Seasonal Diarrhetic Shellfish Toxin Contamination" Toxins 13, no. 11: 784. https://doi.org/10.3390/toxins13110784