ClbG in Avian Pathogenic Escherichia coli Contributes to Meningitis Development in a Mouse Model



 ,
, 
Abstract
:1. Introduction
2. Results
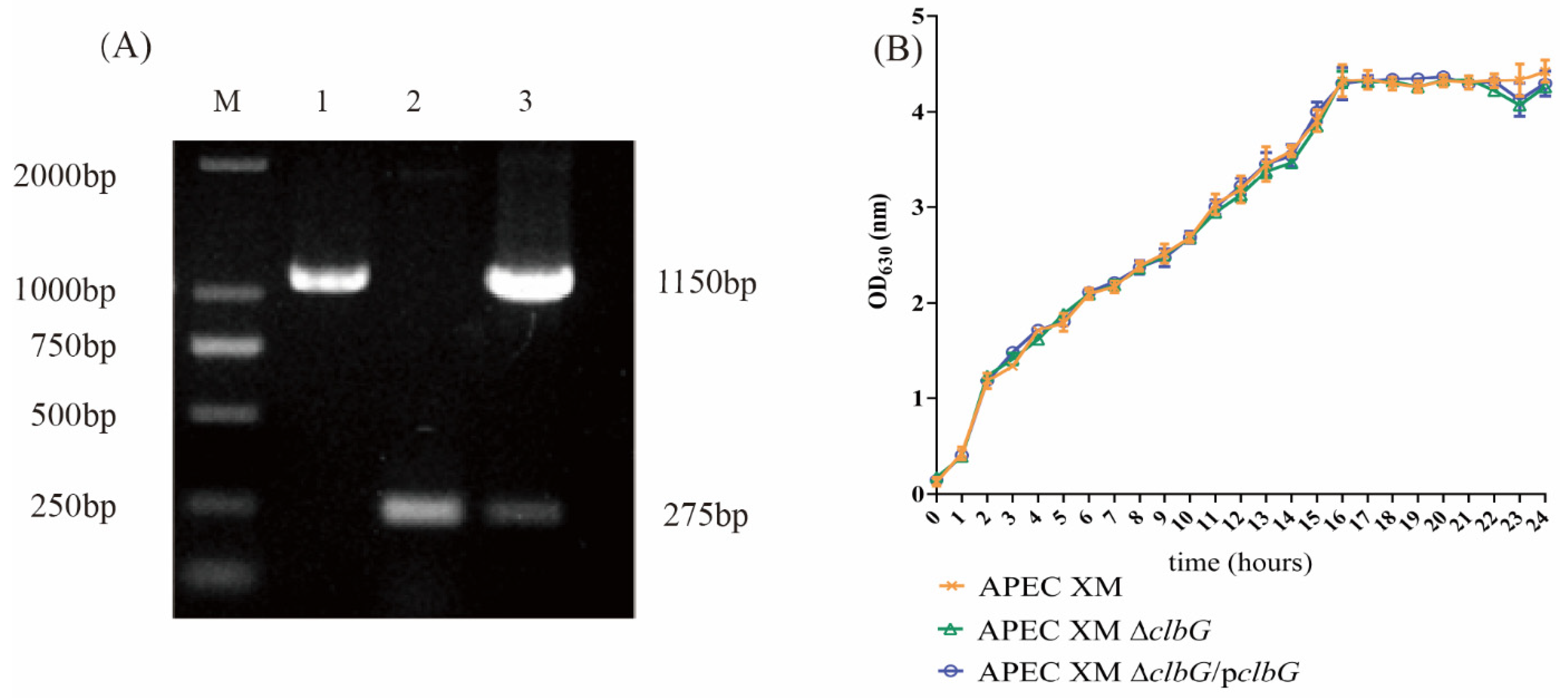
2.1. Genetic Stability and Growth Curves of the Deletion and Complemented Mutants
2.2. Deletion of clbG Affects the Colibactin Production of Avian Pathogenic Escherichia coli (APEC) XM
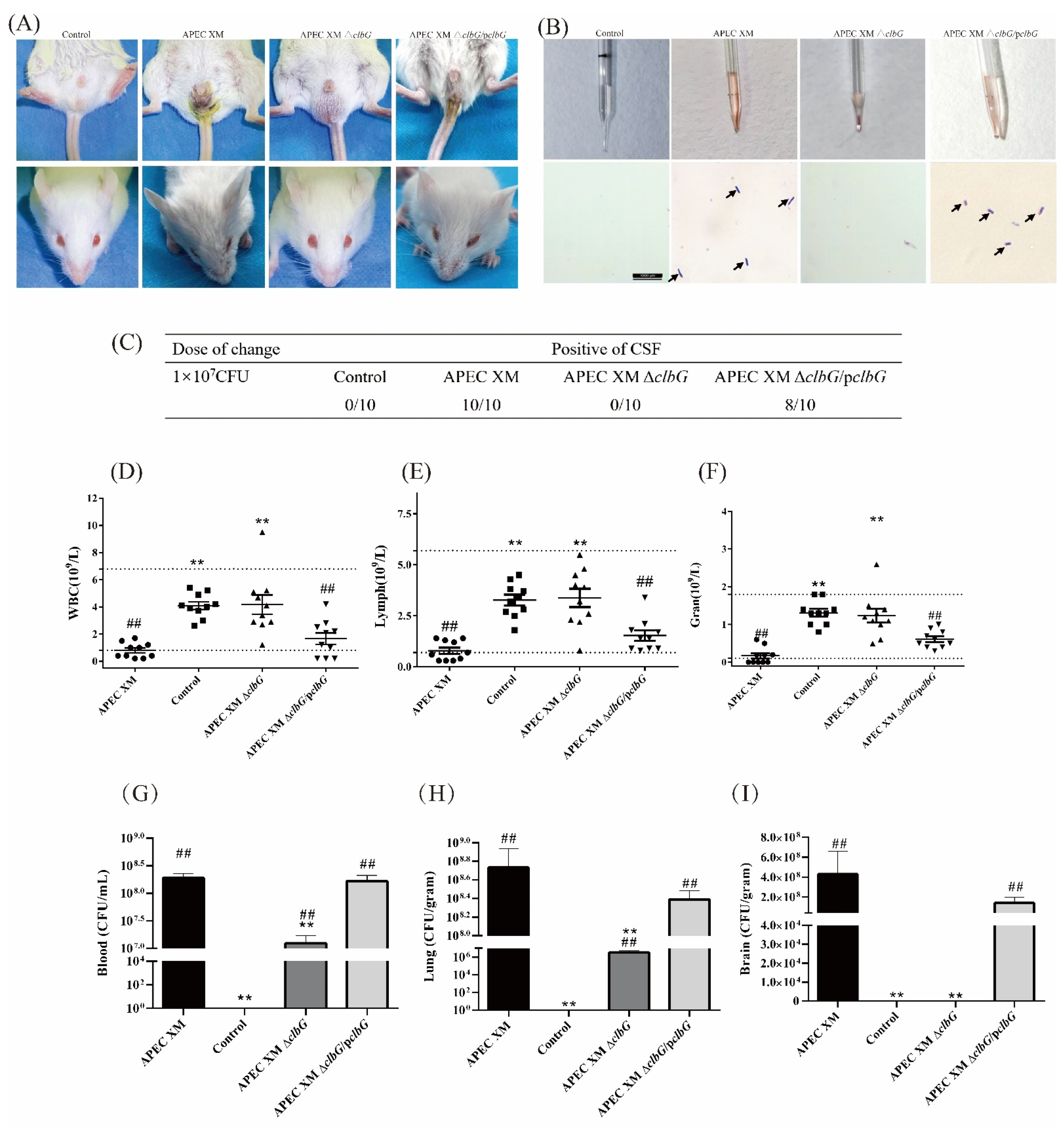
2.3. ClbG Is Required for the Pathogenicity of APEC XM In Vivo
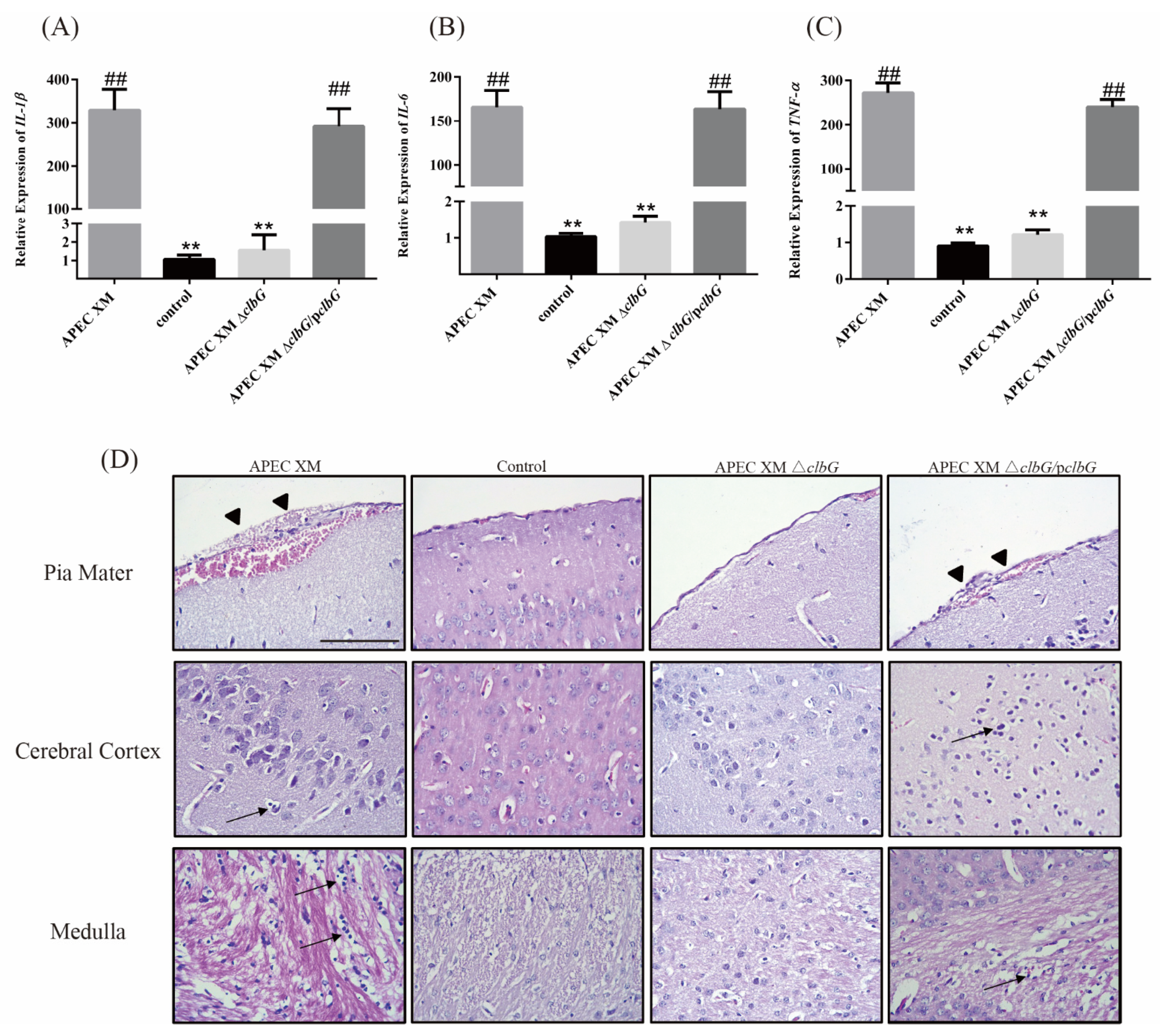
2.4. Relative Cytokines Profiles and Histopathological Findings in Mouse Brains
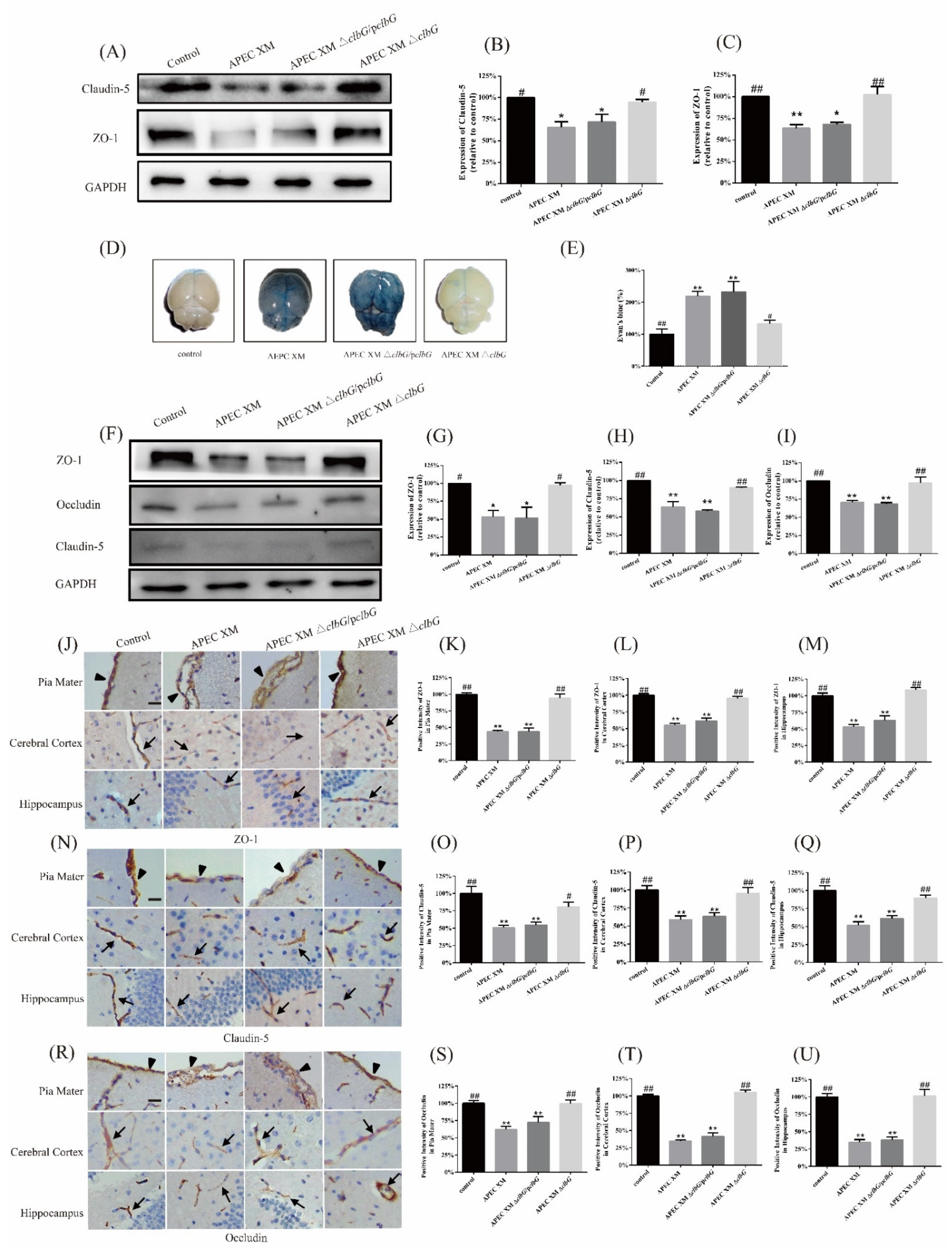
2.5. ClbG Contributes to the Disruption of the Blood–Brain Barrier In Vitro and In Vivo
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Ethics Statement
5.2. Bacterial Strains, Growth Conditions, and Plasmids
5.3. Construction of clbG Deletion and Complemented Mutants
5.4. Growth Curves
5.5. Cell Culture
5.6. Colibactin Cytotoxicity Assays
5.7. E. coli Meningitis Mouse Model
5.8. Evans Blue (EB) Permeability Assay
5.9. Determination of Bacteria Loadings in the Blood, Lung, and Brain
5.10. Histopathology of the Brain
5.11. Immunohistochemistry of Tight Junction Proteins
5.12. IL-1β, IL-6, and TNF-α mRNA Expression in Brains
5.13. Western Blotting
5.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nougayrede, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Putze, J.; Hennequin, C.; Nougayrède, J.P.; Zhang, W.; Homburg, S.; Karch, H.; Bringer, M.A.; Fayolle, C.; Carniel, E.; Rabsch, W.; et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 2009, 77, 4696–4703. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Meng, X.; Li, J.; Chen, Y.; Zhang, D.; Zhong, H.; Xia, P.; Cui, L.; Zhu, G.; Wang, H. Transcriptome profiling of avian pathogenic Escherichia coli and the mouse microvascular endothelial cell line bEnd.3 during interaction. PeerJ 2020, 8, e9172. [Google Scholar] [CrossRef] [PubMed]
- Secher, T.; Samba-Louaka, A.; Oswald, E.; Nougayrède, J.P. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS ONE 2013, 8, e77157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payros, D.; Secher, T.; Boury, M.; Brehin, C.; Ménard, S.; Salvador-Cartier, C.; Cuevas-Ramos, G.; Watrin, C.; Marcq, I.; Nougayrède, J.P.; et al. Maternally acquired genotoxic Escherichia coli alters offspring’s intestinal homeostasis. Gut Microbes 2014, 5, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Pathogenesis of bacterial meningitis: From bacteraemia to neuronal injury. Nat. Rev. Neurosci. 2003, 4, 376–385. [Google Scholar] [CrossRef]
- Ewers, C.; Antao, E.M.; Diehl, I.; Philipp, H.C.; Wieler, L.H. Intestine and environment of the chicken as reservoirs for extraintestinal pathogenic Escherichia coli strains with zoonotic potential. Appl. Environ. Microbiol. 2009, 75, 184–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulin-Schouleur, M.; Reperant, M.; Laurent, S.; Bree, A.; Mignon-Grasteau, S.; Germon, P.; Rasschaert, D.; Schouler, C. Extraintestinal pathogenic Escherichia coli strains of avian and human origin: Link between phylogenetic relationships and common virulence patterns. J. Clin. Microbiol. 2007, 45, 3366–3376. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R.; Oswald, E.; O’Bryan, T.T.; Kuskowski, M.A.; Spanjaard, L. Phylogenetic distribution of virulence-associated genes among Escherichia coli isolates associated with neonatal bacterial meningitis in the Netherlands. J. Infect. Dis. 2002, 185, 774–784. [Google Scholar] [CrossRef] [Green Version]
- Bonacorsi, S.; Bingen, E. Molecular epidemiology of Escherichia coli causing neonatal meningitis. Int. J. Med. Microbiol. 2005, 295, 373–381. [Google Scholar] [CrossRef]
- Saha, O.; Hoque, M.N.; Islam, O.K.; Rahaman, M.M.; Sultana, M.; Hossain, M.A. Multidrug-resistant Avian Pathogenic Escherichia coli strains and association of their virulence genes in Bangladesh. Microorganisms 2020, 8, 1135. [Google Scholar] [CrossRef]
- Rahayuningtyas, I.; Indrawati, A.; Wibawan, I.W.T.; Palupi, M.F.; Istiyaningsih, I. Phylogenetic group determination and plasmid virulence gene profiles of colistin-resistant Escherichia coli originated from the broiler meat supply chain in Bogor, Indonesia. Vet. World 2020, 13, 1807–1814. [Google Scholar] [CrossRef]
- Nielsen, D.W.; Ricker, N.; Barbieri, N.L.; Allen, H.K.; Nolan, L.K.; Logue, C.M. Outer membrane protein A (OmpA) of extraintestinal pathogenic Escherichia coli. BMC Res. Notes 2020, 13, 51. [Google Scholar] [CrossRef]
- Mitchell, N.M.; Johnson, J.R.; Johnston, B.; Curtiss, R., 3rd; Mellata, M. Zoonotic potential of Escherichia coli isolates from retail chicken meat products and eggs. Appl. Environ. Microbiol. 2015, 81, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.; Chang, A.C.; Hodges, J.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Nicholson, B.A.; Nolan, L.K.; Prasadarao, N.V. Serotype O18 avian pathogenic and neonatal meningitis Escherichia coli strains employ similar pathogenic strategies for the onset of meningitis. Virulence 2015, 6, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Tivendale, K.A.; Logue, C.M.; Kariyawasam, S.; Jordan, D.; Hussein, A.; Li, G.; Wannemuehler, Y.; Nolan, L.K. Avian-pathogenic Escherichia coli strains are similar to neonatal meningitis E. coli strains and are able to cause meningitis in the rat model of human disease. Infect. Immun. 2010, 78, 3412–3419. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Siek, K.E.; Giddings, C.W.; Doetkott, C.; Johnson, T.J.; Fakhr, M.K.; Nolan, L.K. Comparison of Escherichia coli isolates implicated in human urinary tract infection and avian colibacillosis. J. Gen. Microbiol. 2005, 151, 2097–2110. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.J.; Kariyawasam, S.; Wannemuehler, Y.; Mangiamele, P.; Johnson, S.J.; Doetkott, C.; Skyberg, J.A.; Lynne, A.M.; Johnson, J.R.; Nolan, L.K. The genome sequence of avian pathogenic Escherichia coli strain O1:K1:H7 shares strong similarities with human extraintestinal pathogenic E. coli genomes. J. Bacteriol. 2007, 189, 3228–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowrouzian, F.L.; Oswald, E. Escherichia coli strains with the capacity for long-term persistence in the bowel microbiota carry the potentially genotoxic pks island. Microb. Pathog. 2012, 53, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Johnston, B.; Kuskowski, M.A.; Nougayrede, J.P.; Oswald, E. Molecular epidemiology and phylogenetic distribution of the Escherichia coli pks genomic island. J. Clin. Microbiol. 2008, 46, 3906–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, D.; Delmas, J.; Cady, A.; Robin, F.; Sivignon, A.; Oswald, E.; Bonnet, R. Cyclomodulins in urosepsis strains of Escherichia coli. J. Clin. Microbiol. 2010, 48, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, A.J.; Martin, P.; Cloup, E.; Stabler, R.A.; Oswald, E.; Taylor, P.W. The Genotoxin Colibactin Is a Determinant of Virulence in Escherichia coli K1 Experimental Neonatal Systemic Infection. Infect. Immun. 2015, 83, 3704–3711. [Google Scholar] [CrossRef] [Green Version]
- Wijetunge, D.S.; Gongati, S.; DebRoy, C.; Kim, K.S.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Kariyawasam, S. Characterizing the pathotype of neonatal meningitis causing Escherichia coli (NMEC). BMC Microbiol. 2015, 15, 211. [Google Scholar] [CrossRef] [Green Version]
- Logue, C.M.; Doetkott, C.; Mangiamele, P.; Wannemuehler, Y.M.; Johnson, T.J.; Tivendale, K.A.; Li, G.; Sherwood, J.S.; Nolan, L.K. Genotypic and phenotypic traits that distinguish neonatal meningitis-associated Escherichia coli from fecal E. coli isolates of healthy human hosts. Appl. Environ. Microbiol. 2012, 78, 5824–5830. [Google Scholar] [CrossRef] [Green Version]
- Marcq, I.; Martin, P.; Payros, D.; Cuevas-Ramos, G.; Boury, M.; Watrin, C.; Nougayrede, J.P.; Olier, M.; Oswald, E. The genotoxin colibactin exacerbates lymphopenia and decreases survival rate in mice infected with septicemic Escherichia coli. J. Infect. Dis. 2014, 210, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.C.; Chen, Y.T.; Chiang, M.K.; Wang, Y.C.; Hsiao, P.Y.; Huang, Y.J.; Lin, C.T.; Cheng, C.C.; Liang, C.L.; Lai, Y.C. Colibactin Contributes to the Hypervirulence of pks(+) K1 CC23 Klebsiella pneumoniae in Mouse Meningitis Infections. Front. Cell. Infect. Microbiol. 2017, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, A.R.; Wernke, K.M.; Kim, C.S.; Lees, N.R.; Crawford, J.M.; Herzon, S.B. Synthesis and reactivity of precolibactin 886. Nat. Chem. 2019, 11, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, A.O.; Garcie, C.; Wu, V.; Martin, P.; Ueoka, R.; Oswald, E.; Piel, J. Colibactin biosynthesis and biological activity depend on the rare aminomalonyl polyketide precursor. Chem. Commun. 2015, 51, 13138–13141. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.; Raivich, G.; Wellmer, A.; Noeske, C.; Kunst, T.; Werner, A.; Brück, W.; Nau, R. A mouse model of Streptococcus pneumoniae meningitis mimicking several features of human disease. Acta Neuropathol. 2001, 101, 499–508. [Google Scholar] [CrossRef]
- Ribes, S.; Regen, T.; Meister, T.; Tauber, S.C.; Schütze, S.; Mildner, A.; Mack, M.; Hanisch, U.K.; Nau, R. Resistance of the brain to Escherichia coli K1 infection depends on MyD88 signaling and the contribution of neutrophils and monocytes. Infect. Immun. 2013, 81, 1810–1819. [Google Scholar] [CrossRef] [Green Version]
- Tauzin, M.; Ouldali, N.; Levy, C.; Bechet, S.; Cohen, R.; Caeymaex, L. Combination therapy with ciprofloxacin and third-generation cephalosporin versus third-generation cephalosporin monotherapy in Escherichia coli meningitis in infants: A multicentre propensity score-matched observational study. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2019, 25, 1006–1012. [Google Scholar] [CrossRef]
- Kim, K.S. Acute bacterial meningitis in infants and children. Lancet Infect. Dis. 2010, 10, 32–42. [Google Scholar] [CrossRef]
- Kim, K.S. Human meningitis-associated Escherichia coli. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewers, C.; Li, G.; Wilking, H.; Kiessling, S.; Alt, K.; Antao, E.M.; Laturnus, C.; Diehl, I.; Glodde, S.; Homeier, T.; et al. Avian pathogenic, uropathogenic, and newborn meningitis-causing Escherichia coli: How closely related are they? Int. J. Med. Microbiol. 2007, 297, 163–176. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Frisan, T.; Mihaljevic, B. Bacterial Genotoxins as the Interphase Between DNA Damage and Immune Response. In Microbial Toxins; Stiles, B., Alape-Girón, A., Dubreuil, J.D., Mandal, M., Gopalakrishnakone, P., Eds.; Springer: Dordrecht, The Netherlands, 2018; pp. 383–402. [Google Scholar]
- Zha, L.; Wilson, M.R.; Brotherton, C.A.; Balskus, E.P. Characterization of Polyketide Synthase Machinery from the pks Island Facilitates Isolation of a Candidate Precolibactin. ACS Chem. Biol. 2016, 11, 1287–1295. [Google Scholar] [CrossRef]
- Dietzman, D.E.; Fischer, G.W.; Schoenknecht, F.D. Neonatal Escherichia coli septicemia--bacterial counts in blood. J. Pediatrics 1974, 85, 128–130. [Google Scholar] [CrossRef]
- Kim, K.S.; Itabashi, H.; Gemski, P.; Sadoff, J.; Warren, R.L.; Cross, A.S. The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J. Clin. Investig. 1992, 90, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Secher, T.; Brehin, C.; Oswald, E. Early settlers: Which, E. coli strains do you not want at birth? Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G123–G129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secher, T.; Payros, D.; Brehin, C.; Boury, M.; Watrin, C.; Gillet, M.; Bernard-Cadenat, I.; Menard, S.; Theodorou, V.; Saoudi, A.; et al. Oral tolerance failure upon neonatal gut colonization with Escherichia coli producing the genotoxin colibactin. Infect. Immun. 2015, 83, 2420–2429. [Google Scholar] [CrossRef] [Green Version]
- Pfister, H.W.; Fontana, A.; Tauber, M.G.; Tomasz, A.; Scheld, W.M. Mechanisms of brain injury in bacterial meningitis: Workshop summary. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1994, 19, 463–479. [Google Scholar] [CrossRef] [Green Version]
- Nau, R.; Soto, A.; Bruck, W. Apoptosis of neurons in the dentate gyrus in humans suffering from bacterial meningitis. J. Neuropathol. Exp. Neurol. 1999, 58, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheld, W.M.; Koedel, U.; Nathan, B.; Pfister, H.W. Pathophysiology of bacterial meningitis: Mechanism(s) of neuronal injury. J. Infect. Dis. 2002, 186 (Suppl. 2), S225–S233. [Google Scholar] [CrossRef]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Winkler, F.; Koedel, U.; Kastenbauer, S.; Pfister, H.W. Differential expression of nitric oxide synthases in bacterial meningitis: Role of the inducible isoform for blood-brain barrier breakdown. J. Infect. Dis. 2001, 183, 1749–1759. [Google Scholar] [CrossRef]
- Koedel, U.; Paul, R.; Winkler, F.; Kastenbauer, S.; Huang, P.L.; Pfister, H.W. Lack of endothelial nitric oxide synthase aggravates murine pneumococcal meningitis. J. Neuropathol. Exp. Neurol. 2001, 60, 1041–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, Z.; Chang, X.; Wang, X.; Hu, J.; Lin, Q.; Jia, Y.; Yang, X.; Wang, X. Disruption of blood-brain barrier by an Escherichia coli isolated from canine septicemia and meningoencephalitis. Comp. Immunol. Microbiol. Infect. Dis. 2019, 63, 44–50. [Google Scholar] [CrossRef]
- Eisenhauer, P.B.; Jacewicz, M.S.; Conn, K.J.; Koul, O.; Wells, J.M.; Fine, R.E.; Newburg, D.S. Escherichia coli Shiga toxin 1 and TNF-alpha induce cytokine release by human cerebral microvascular endothelial cells. Microb. Pathog. 2004, 36, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Chang, A.C.; Stoltz, B.M.; Prasadarao, N.V. Escherichia coli K1 modulates peroxisome proliferator-activated receptor gamma and glucose transporter 1 at the blood-brain barrier in neonatal meningitis. J. Infect. Dis. 2016, 214, 1092–1104. [Google Scholar] [CrossRef] [Green Version]
- Lemon, J.K.; Miller, M.R.; Weiser, J.N. Sensing of interleukin-1 cytokines during Streptococcus pneumoniae colonization contributes to macrophage recruitment and bacterial clearance. Infect. Immun. 2015, 83, 3204–3212. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.L.; Shi, F.D.; Zhou, Q.; Liu, Q.Y.; Wang, Y.X.; Song, Y.; Wu, Z.S.; Shi, Y.H.; Zhang, L.; Xu, K.Z.; et al. Interleukin-1beta protection against experimental sepsis in mice. Inflammation 2021, 44, 358–370. [Google Scholar] [CrossRef]
- Merrill, J.E.; Benveniste, E.N. Cytokines in inflammatory brain lesions: Helpful and harmful. Trends Neurosci. 1996, 19, 331–338. [Google Scholar] [CrossRef]
- Benveniste, E.N. Cytokines: Influence on glial cell gene expression and function. Chem. Immunol. 1992, 52, 106–153. [Google Scholar] [PubMed]
- Furth, A.M.V.; Roord, J.J.; Furth, R.V. Roles of proinflammatory and anti-inflammatory cytokines in pathophysiology of bacterial meningitis and effect of adjunctive therapy-15. Infect. Immun. 1996, 64, 4883–4890. [Google Scholar] [CrossRef] [Green Version]
- Wellmer, A.; Gerber, J.; Ragheb, J.; Zysk, G.; Kunst, T.; Smirnov, A.; Bruck, W.; Nau, R. Effect of deficiency of tumor necrosis factor alpha or both of its receptors on Streptococcus pneumoniae central nervous system infection and peritonitis. Infect. Immun. 2001, 69, 6881–6886. [Google Scholar] [CrossRef] [Green Version]
- Damas, P.; Ledoux, D.; Nys, M.; Vrindts, Y.; De Groote, D.; Franchimont, P.; Lamy, M. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann. Surg. 1992, 215, 356–362. [Google Scholar] [CrossRef]
- Voirin, A.C.; Perek, N.; Roche, F. Inflammatory stress induced by a combination of cytokines (IL-6, IL-17, TNF-alpha) leads to a loss of integrity on bEnd.3 endothelial cells in vitro BBB model. Brain Res. 2020, 1730, 146647. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Zhong, W.H.; Song, W.L.; Deng, Y.Y.; Yang, D.M.; Xiong, B.; Zeng, H.K.; Wang, H.D. Ulinastatin ameliorates pulmonary capillary endothelial permeability induced by sepsis through protection of tight junctions via inhibition of TNF-alpha and related pathways. Front. Pharmacol. 2018, 9, 823. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, X.; Miao, C.; Chen, J. Propofol attenuated TNF-α-modulated occludin expression by inhibiting Hif-1α/VEGF/VEGFR-2/ERK signaling pathway in hCMEC/D3 cells. BMC Anesthesiol. 2019, 19, 127. [Google Scholar] [CrossRef] [Green Version]
- Clark, P.R.; Kim, R.K.; Pober, J.S.; Kluger, M.S. Tumor necrosis factor disrupts claudin-5 endothelial tight junction barriers in two distinct NF-kappaB-dependent phases. PLoS ONE 2015, 10, e0120075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Hashimoto, K.; Go, H.; Miyazaki, K.; Sato, M.; Kawasaki, Y.; Momoi, N.; Hosoya, M. Towards the development of a human in vitro model of the blood-brain barrier for virus-associated acute encephalopathy: Assessment of the time- and concentration-dependent effects of TNF-alpha on paracellular tightness. Exp. Brain Res. 2021, 239, 451–461. [Google Scholar] [CrossRef]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef]
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of blood brain barrier disruption by different types of bacteria, and bacterial-host interactions facilitate the bacterial pathogen invading the brain. Cell. Mol. Neurobiol. 2018, 38, 1349–1368. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.-C.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You Shall Not Pass”—Tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Hancock, B.M.; Bermudez, A.; Del Cid, N.; Reyes, E.; van Sorge, N.M.; Lauth, X.; Smurthwaite, C.A.; Hilton, B.J.; Stotland, A.; et al. Bacterial induction of Snail1 contributes to blood-brain barrier disruption. J. Clin. Investig. 2015, 125, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, A.; Rochfort, K.D.; McDonnell, C.J.; Kerrigan, S.W.; Cummins, P.M. Staphylococcus aureus-mediated blood—Brain barrier injury: An in vitro human brain microvascular endothelial cell model. Cell. Microbiol. 2017, 19, e12664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.J.; Bee, O.B.; McDonagh, M.A.; Stebbins, M.J.; Palecek, S.P.; Doran, K.S.; Shusta, E.V. Modeling Group B Streptococcus and blood-brain barrier interaction by using induced pluripotent stem cell-derived brain endothelial cells. mSphere 2017, 2, e00398-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins Gomes, S.F.; Westermann, A.J.; Sauerwein, T.; Hertlein, T.; Forstner, K.U.; Ohlsen, K.; Metzger, M.; Shusta, E.V.; Kim, B.J.; Appelt-Menzel, A.; et al. Induced pluripotent stem cell-derived brain endothelial cells as a cellular model to study Neisseria meningitidis infection. Front. Microbiol. 2019, 10, 1181. [Google Scholar] [CrossRef]
- Tenenbaum, T.; Matalon, D.; Adam, R.; Seibt, A.; Wewer, C.; Schwerk, C.; Galla, H.J.; Schroten, H. Dexamethasone prevents alteration of tight junction-associated proteins and barrier function in porcine choroid plexus epithelial cells after infection with Streptococcus suis in vitro. Brain Res. 2008, 1229, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cai, M.; Hu, J.; Zhang, Z.; Wang, X.; Chang, X.; Zhang, F.; Guo, C.; Wang, X. Mechanism of blood-brain barrier disruption by an Escherichia coli from lambs with severe diarrhea and meningoencephalitis. Microb. Pathog. 2020, 147, 104288. [Google Scholar] [CrossRef]
- Stevenson, B.R.; Siliciano, J.D.; Mooseker, M.S.; Goodenough, D.A. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol. 1986, 103, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Stamatovic, S.M.; Sladojevic, N.; Keep, R.F.; Andjelkovic, A.V. PDCD10 (CCM3) regulates brain endothelial barrier integrity in cerebral cavernous malformation type 3: Role of CCM3-ERK1/2-cortactin cross-talk. Acta Neuropathol. 2015, 130, 731–750. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.T.; Burgers, H.F.; Rabie, T.; Marti, H.H. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J. Cereb. Blood Flow Metab. 2010, 30, 837–848. [Google Scholar] [CrossRef] [Green Version]
- DeMaio, L.; Rouhanizadeh, M.; Reddy, S.; Sevanian, A.; Hwang, J.; Hsiai, T.K. Oxidized phospholipids mediate occludin expression and phosphorylation in vascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H674–H683. [Google Scholar] [CrossRef]
- Kabra, R.; Knight, K.K.; Zhou, R.; Snyder, P.M. Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J. Biol. Chem. 2008, 283, 6033–6039. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Teng, T.; Li, R.; Simonyi, A.; Sun, G.Y.; Lee, J.C. TNFalpha alters occludin and cerebral endothelial permeability: Role of p38MAPK. PLoS ONE 2017, 12, e0170346. [Google Scholar] [CrossRef]
- Morita, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J. Cell Biol. 1999, 147, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgintino, D.; Errede, M.; Robertson, D.; Capobianco, C.; Girolamo, F.; Vimercati, A.; Bertossi, M.; Roncali, L. Immunolocalization of tight junction proteins in the adult and developing human brain. Histochem. Cell Biol. 2004, 122, 51–59. [Google Scholar] [CrossRef]
- Wen, H.; Watry, D.D.; Marcondes, M.C.; Fox, H.S. Selective decrease in paracellular conductance of tight junctions: Role of the first extracellular domain of claudin-5. Mol. Cell. Biol. 2004, 24, 8408–8417. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, M.C.; Saadoun, S.; Binder, D.K.; Manley, G.T.; Krishna, S.; Verkman, A.S. Molecular mechanisms of brain tumor edema. Neuroscience 2004, 129, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, W.; Piontek, A.; Protze, J.; Eichner, M.; Mahringer, A.; Subileau, E.A.; Lee, I.M.; Schulzke, J.D.; Krause, G.; Piontek, J. Reversible opening of the blood-brain barrier by claudin-5-binding variants of Clostridium perfringens enterotoxin’s claudin-binding domain. Biomaterials 2018, 161, 129–143. [Google Scholar] [CrossRef]
- Platania, C.B.M.; Lazzara, F.; Fidilio, A.; Fresta, C.G.; Conti, F.; Giurdanella, G.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. Blood-retinal barrier protection against high glucose damage: The role of P2X7 receptor. Biochem. Pharmacol. 2019, 168, 249–258. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Marcq, I.; Magistro, G.; Penary, M.; Oswald, E. Interplay between Siderophores and Colibactin Genotoxin Biosynthetic Pathways in Escherichia coli. PLoS Pathog. 2013, 9, e1003437. [Google Scholar] [CrossRef]
- Solovjeva, L.; Firsanov, D.; Pleskach, N.; Svetlova, M. Immunofluorescence Analysis of γ-H2AX Foci in Mammalian Fibroblasts at Different Phases of the Cell Cycle. Methods Mol. Biol. 2017, 1644, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Hejair, H.M.A.; Ma, J.; Zhu, Y.; Sun, M.; Dong, W.; Zhang, Y.; Pan, Z.; Zhang, W.; Yao, H. Role of outer membrane protein T in pathogenicity of avian pathogenic Escherichia coli. Res. Vet. Sci. 2017, 115, 109–116. [Google Scholar] [CrossRef]
- Manaenko, A.; Chen, H.; Kammer, J.; Zhang, J.H.; Tang, J. Comparison Evans Blue injection routes: Intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. J. Neurosci. Methods 2011, 195, 206–210. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain or Plasmid | Characteristic or Function | Source |
|---|---|---|
| Strains | ||
| Avian pathogenic Escherichia coli (APEC) XM | Virulent strain of APEC | Donated by Dr. Guoqiang Zhu, Yangzhou University |
| APEC XM ΔclbG | Deletion mutant of clbG with APEC XM background | This study |
| APEC XM ΔclbG/pclbG | APEC-XM ΔclbG with the vector pACYC184-clbG, Cmr | This study |
| Plasmid | ||
| pKD46 | λ red recombinase expression plasmid | [90] |
| pKD3 | pANTSγ derivative containing FRT-flanked, Cmr | [90] |
| pCP20 | temperature-sensitive replication and thermal induction of FLP synthesis | [90] |
| pACYC184-clbG | pACYC184 containing the promoter followed by the full-length clbG, Cmr | This study |
| Sequence (5′→3′) | Product size | |
| P1 | GTCCTCTTCGCTGGATGT | 1150/275 |
| P2 | GAACATCAGTGCGACATC | |
| P3 | TGCGCACTGGCAGCCACATCGGCGGCGCGGTGATGGCGTGTGGCTGTCTGTGTAGGCTGGAGCTGCTTC | 1000 |
| P4 | GCTCCGGTTCGCAATATGTAGGCATGGCACGGTGGCTGTATGAGCGTTCATATGAATATCCTCCTTAG | |
| P5 | CTAACGCAGTCAGGCACCGTGTATGACGAAGGATGTCGCACTGATG | 1150 |
| P6 | GTGCCGCCGGCTTCCATTTACGCC TGTCCGCCGTTG | |
| GAPDH | AACGGGAAGCCCATCACCATC | 98 |
| AAGACACCAGTAGACTCCACGA | ||
| IL-1β | ATGAAAGACGGCACACCCAC | 175 |
| GCTTGTGCTCTGCTTGTGAG | ||
| IL-6 | TGCAAGAGACTTCCATCCAGT | 71 |
| GTGAAGTAGGGAAGGCCG | ||
| TNF-α | ACTGAACTTCGGGGTGATCG | 97 |
| TGATCTGAGTGTGAGGGTCTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Zhang, J.; Chen, Y.; Zhong, H.; Wang, H.; Li, J.; Zhu, G.; Xia, P.; Cui, L.; Li, J.; et al. ClbG in Avian Pathogenic Escherichia coli Contributes to Meningitis Development in a Mouse Model. Toxins 2021, 13, 546. https://doi.org/10.3390/toxins13080546
Wang P, Zhang J, Chen Y, Zhong H, Wang H, Li J, Zhu G, Xia P, Cui L, Li J, et al. ClbG in Avian Pathogenic Escherichia coli Contributes to Meningitis Development in a Mouse Model. Toxins. 2021; 13(8):546. https://doi.org/10.3390/toxins13080546
Chicago/Turabian StyleWang, Peili, Jiaxiang Zhang, Yanfei Chen, Haoran Zhong, Heng Wang, Jianji Li, Guoqiang Zhu, Pengpeng Xia, Luying Cui, Jun Li, and et al. 2021. "ClbG in Avian Pathogenic Escherichia coli Contributes to Meningitis Development in a Mouse Model" Toxins 13, no. 8: 546. https://doi.org/10.3390/toxins13080546