Engineering and Structural Insights of a Novel BBI-like Protease Inhibitor Livisin from the Frog Skin Secretion

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
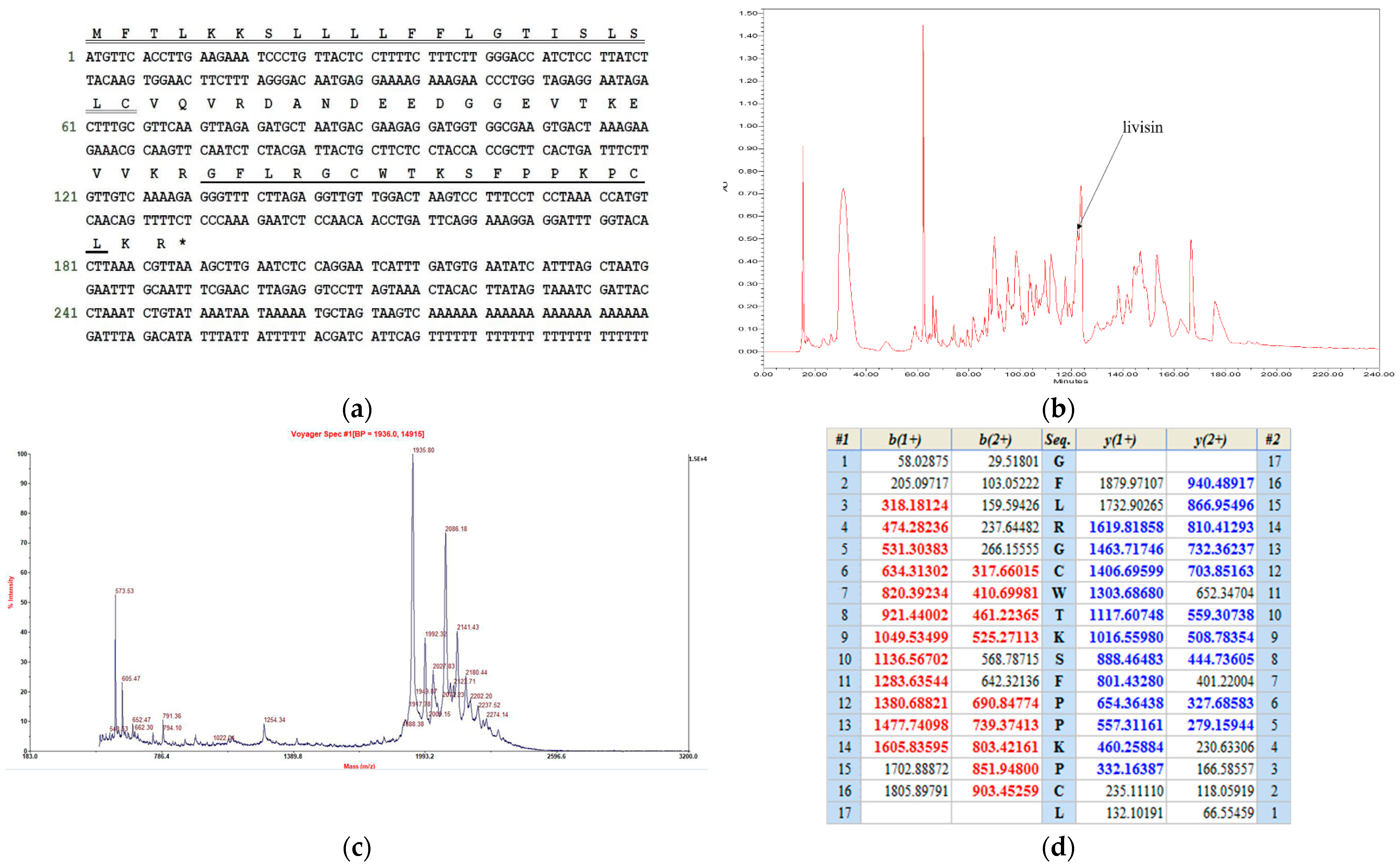
2.1. Cloning of the Livisin Precursor-Encoding cDNA and Structural Characterization of Livisin
2.2. Design, Synthesis, and Secondary Structure Confirmation of Livisin and Analogs
2.2.1. Design and Synthesis of the Native Livisin and Analogs
2.2.2. Confirmation of the Secondary Structures of Native Livisin and Analogs by Circular Dichroism
2.3. Effects of Synthetic Livisin and Its Analogs on Protease Inhibition
2.4. Antimicrobial/Hemolytic Activities of the Peptides
2.5. Molecular Docking and Structure–Activity Relationships of Livisin and the Analogs
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Zhou, M.; Chen, W.; Chen, T.; Walker, B.; Shaw, C. HV-BBI-A novel amphibian skin Bowman-Birk-like trypsin inhibitor. Biochem. Biophys. Res. Commun. 2008, 372, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Kim, J.B. A protease inhibitor of the Kunitz family from skin secretions of the tornato frog, Dyscophus guineti (Microhylidae). Biochem. Biophys. Res. Commun. 2000, 279, 961–964. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Zhou, M.; Yang, M.; Ma, C.; Chen, T.; Zhang, Y.; Zeller, M.; Hornshaw, M.; Shaw, C. Functional peptidomics of amphibian skin secretion: A novel Kunitz-type chymotrypsin inhibitor from the African hyperoliid frog, Kassina senegalensis. Biochimie 2012, 94, 891–899. [Google Scholar] [CrossRef]
- Wang, M.; Wang, L.; Chen, T.; Walker, B.; Zhou, M.; Sui, D.; Conlon, J.M.; Shaw, C. Identification and molecular cloning of a novel amphibian Bowman Birk-type trypsin inhibitor from the skin of the Hejiang Odorous Frog; Odorrana hejiangensis. Peptides 2012, 33, 245–250. [Google Scholar] [CrossRef]
- Lin, Y.; Hang, H.; Chen, T.; Zhou, M.; Wang, L.; Shaw, C. PLR-HL: A Novel Amphibian Bowman-Birk-type Trypsin Inhibitor from the Skin Secretion of the Broad-folded Frog, Hylarana latouchii. Chem. Biol. Drug Des. 2016, 87, 91–100. [Google Scholar] [CrossRef]
- Safavi, F.; Rostami, A. Role of serine proteases in inflammation: Bowman-Birk protease inhibitor (BBI) as a potential therapy for autoimmune diseases. Exp. Mol. Pathol. 2012, 93, 428–433. [Google Scholar] [CrossRef]
- Losso, J.N. The biochemical and functional food properties of the Bowman-Birk inhibitor. Crit. Rev. Food Sci. Nutr. 2008, 48, 94–118. [Google Scholar] [CrossRef]
- McBride, J.D.; Watson, E.M.; Brauer, A.B.E.; Jaulent, A.M.; Leatherbarrow, R.J. Peptide mimics of the Bowman-Birk inhibitor reactive site loop. Biopolymers 2002, 66, 79–92. [Google Scholar] [CrossRef]
- Rogers, D.M.; Jasim, S.B.; Dyer, N.T.; Auvray, F.; Réfrégiers, M.; Hirst, J.D. Electronic Circular Dichroism Spectroscopy of Proteins. Chem 2019, 5, 2751–2774. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- De Veer, S.J.; Li, C.Y.; Swedberg, J.E.; Schroeder, C.I.; Craik, D.J. Engineering potent mesotrypsin inhibitors based on the plant-derived cyclic peptide, sunflower trypsin inhibitor-1. Eur. J. Med. Chem. 2018, 155, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Cotabarren, J.; Broitman, D.J.; Quiroga, E.; Obregón, W.D. GdTI, the first thermostable trypsin inhibitor from Geoffroea decorticans seeds. A novel natural drug with potential application in biomedicine. Int. J. Biol. Macromol. 2020, 148, 869–879. [Google Scholar] [CrossRef]
- Cotabarren, J.; Lufrano, D.; Parisi, M.G.; Obregón, W.D. Biotechnological, biomedical, and agronomical applications of plant protease inhibitors with high stability: A systematic review. Plant Sci. 2020, 292, 110398. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, T.; Walker, B.; Shaw, C. Lividins: Novel antimicrobial peptide homologs from the skin secretion of the Chinese Large Odorous frog, Rana (Odorrana) livida. Identification by “shotgun” cDNA cloning and sequence analysis. Peptides 2006, 27, 2118–2123. [Google Scholar] [CrossRef]
- Vanhoye, D.; Bruston, F.; Nicolas, P.; Amiche, M. Antimicrobial peptides from hylid and ranin frogs originated from a 150-million-year-old ancestral precursor with a conserved signal peptide but a hypermutable antimicrobial domain. Eur. J. Biochem. 2003, 270, 2068–2081. [Google Scholar] [CrossRef]
- Xu, X.; Lai, R. The chemistry and biological activities of peptides from amphibian skin secretions. Chem. Rev. 2015, 115, 1760–1846. [Google Scholar] [CrossRef]
- De Veer, S.J.; Wang, C.K.; Harris, J.M.; Craik, D.J.; Swedberg, J.E. Improving the Selectivity of Engineered Protease Inhibitors: Optimizing the P2 Prime Residue Using a Versatile Cyclic Peptide Library. J. Med. Chem. 2015, 58, 8257–8268. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Chen, G.; Xi, X.; Ma, C.; Wang, L.; Burrows, J.F.; Duan, J.; Zhou, M.; Chen, T. Discovery and rational design of a novel bowman-birk related protease inhibitor. Biomolecules 2019, 9, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, P.; Ge, L.; Ma, R.; Wei, R.; McCrudden, C.M.; Chen, T.; Shaw, C.; Kwok, H.F. Identification and pharmaceutical evaluation of novel frog skin-derived serine proteinase inhibitor peptide–PE-BBI (Pelophylax esculentus Bowman-Birk inhibitor) for the potential treatment of cancer. Sci. Rep. 2018, 8, 14502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, X.; Wu, Y.; Zhou, M.; Ma, C.; Xi, X.; Chen, T.; Walker, B.; Shaw, C.; Wang, L. A Bowman-Birk type chymotrypsin inhibitor peptide from the amphibian, Hylarana erythraea. Sci. Rep. 2018, 8, 5851. [Google Scholar] [CrossRef] [Green Version]
- Grudnik, P.; Debowski, D.; Legowska, A.; Malicki, S.; Golik, P.; Karna, N.; Rolka, K.; Dubin, G. Atomic resolution crystal structure of HV-BBI protease inhibitor from amphibian skin in complex with bovine trypsin. Proteins Struct. Funct. Bioinform. 2015, 83, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Chen, Y.K.; Foley, F.M.; Bansal, P.S.; Bharathi, R.; Clark, R.J.; Sommerhoff, C.P.; Craik, D.J. The absolute structural requirement for a proline in the P3′-position of Bowman-Birk protease inhibitors is surmounted in the minimized SFTI-1 scaffold. J. Biol. Chem. 2006, 281, 23668–23675. [Google Scholar] [CrossRef] [Green Version]
- Sreerama, N.; Manning, M.C.; Powers, M.E.; Zhang, J.X.; Goldenberg, D.P.; Woody, R.W. Tyrosine, phenylalanine, and disulfide contributions to the circular dichroism of proteins: Circular dichroism spectra of wild-type and mutant bovine pancreatic trypsin inhibitor. Biochemistry 1999, 38, 10814–10822. [Google Scholar] [CrossRef]
- Gitlin-Domagalska, A.; Dębowski, D.; Gucwa, K.; Starego, D.; Ptaszyńska, N.; Sieradzan, A.; Karczyńska, A.; Samsonov, S.A.; Mangold, M.; Gütschow, M.; et al. Truncation of Huia versabilis Bowman-Birk inhibitor increases its selectivity, matriptase-1 inhibitory activity and proteolytic stability. Biochimie 2020, 171–172, 178–186. [Google Scholar] [CrossRef]
- Brinckerhoff, L.H.; Kalashnikov, V.V.; Thompson, L.W.; Yamshchikov, G.V.; Pierce, R.A.; Galavotti, H.S.; Engelhard, V.H.; Slingluff, C.L. Terminal modifications inhibit proteolytic degradation of an immunogenic MART-127-35 peptide: Implications for peptide vaccines. Int. J. Cancer 1999, 83, 326–334. [Google Scholar] [CrossRef]
- Dȩbowski, D.; Łukajtis, R.; Łȩgowska, A.; Karna, N.; Pikuła, M.; Wysocka, M.; Maliszewska, I.; Sieńczyk, M.; Lesner, A.; Rolka, K. Inhibitory and antimicrobial activities of OGTI and HV-BBI peptides, fragments and analogs derived from amphibian skin. Peptides 2012, 35, 276–284. [Google Scholar] [CrossRef]
- Tyler, M.J.; Stone, D.J.M.; Bowie, J.H. A novel method for the release and collection of dermal, glandular secretions from the skin of frogs. J. Pharmacol. Toxicol. Methods 1992, 28, 199–200. [Google Scholar] [CrossRef]
- Wu, D.; Gao, Y.; Wang, L.; Xi, X.; Wu, Y.; Zhou, M.; Zhang, Y.; Ma, C.; Chen, T.; Shaw, C. A combined molecular cloning and mass spectrometric method to identify, characterize, and design frenatin peptides from the skin secretion of Litoria infrafrenata. Molecules 2016, 21, 1429. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Österberg, F.; Morris, G.M.; Sanner, M.F.; Olson, A.J.; Goodsell, D.S. Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in autodock. Proteins Struct. Funct. Genet. 2002, 46, 34–40. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock web server—High resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, W249–W253. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhou, C.Y.; Wu, Y.D. Residue-specific force field based on the protein coil library. RSFF1: Modification of OPLS-AA/L. J. Phys. Chem. B 2014, 118, 6983–6998. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Primary Structure 1 | Ki (μM) | ||||
|---|---|---|---|---|---|---|
| Trypsin | Matriptase | Chymotrypsin | Mesotrypsin | 20s Proteasome | ||
| livisin | GFLRGC*WTKSFPPKPC*L | 2.792 | 25.15 | 92.71 | 22.38 | NI 2 |
| livisin-NH2 | GFLRGC*WTKSFPPKPC*L-NH2 | 0.424 | 2.362 | 143.8 | 18.6 | NI |
| livisin-Arg | GFLRGC*WTRSFPPKPC*L | 1.016 | 46.87 | 74.66 | 32.08 | NI |
| livisin-Phe | GFLRGC*WTFSFPPKPC*L | NI | NI | 3.464 | NI | 17.72 |
| livisin-loop | C*WTKSFPPKPC* | 4.252 | NI | NI | 54.85 | NI |
| Strains | Livisin | Livisin-NH2 | Livisin-Arg | Livisin-Phe | Livisin-Loop |
|---|---|---|---|---|---|
| S. aureus (NCTC 10788) | >512 | >512 | >512 | >512 | >512 |
| E. coli (NCTC 10418) | >512 | >512 | >512 | >512 | >512 |
| C. albicans (NCPF 1467) | >512 | 256 | >512 | >512 | >512 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Tong, C.; Qi, J.; Liao, X.; Li, X.; Zhang, X.; Zhou, M.; Wang, L.; Ma, C.; Xi, X.; et al. Engineering and Structural Insights of a Novel BBI-like Protease Inhibitor Livisin from the Frog Skin Secretion. Toxins 2022, 14, 273. https://doi.org/10.3390/toxins14040273
Yang J, Tong C, Qi J, Liao X, Li X, Zhang X, Zhou M, Wang L, Ma C, Xi X, et al. Engineering and Structural Insights of a Novel BBI-like Protease Inhibitor Livisin from the Frog Skin Secretion. Toxins. 2022; 14(4):273. https://doi.org/10.3390/toxins14040273
Chicago/Turabian StyleYang, Jie, Chengliang Tong, Junmei Qi, Xiaoying Liao, Xiaokun Li, Xu Zhang, Mei Zhou, Lei Wang, Chengbang Ma, Xinping Xi, and et al. 2022. "Engineering and Structural Insights of a Novel BBI-like Protease Inhibitor Livisin from the Frog Skin Secretion" Toxins 14, no. 4: 273. https://doi.org/10.3390/toxins14040273