Improving the Annotation of the Venom Gland Transcriptome of Pamphobeteus verdolaga, Prospecting Novel Bioactive Peptides

 , , , , and
, , , , and
Abstract
:
1. Introduction
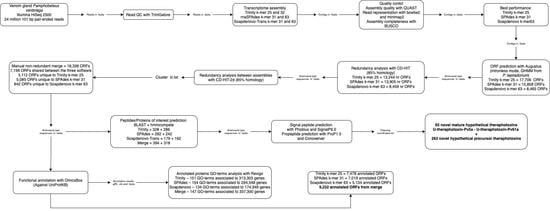
2. Results
2.1. Quality of the Transcripts Assembled with Trinity and SPAdes Are Similar, While Outperforming That of SOAPdenovo-Trans
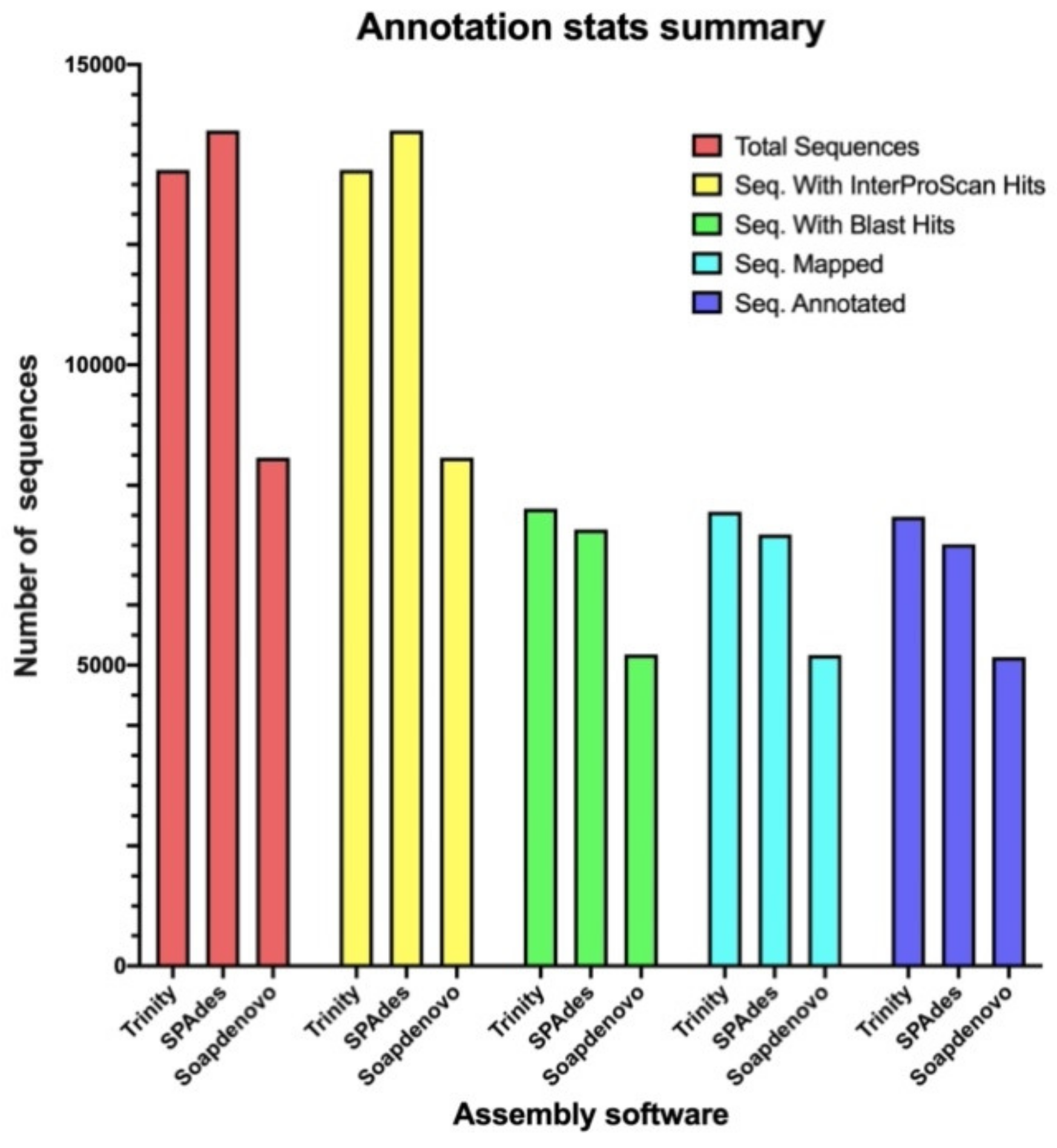
2.2. Trinity, SPAdes, and SOAPdenovo-Trans Assemblies Show Differences in the Quantity of Associated ORFs and the Functionality of the Respective Annotated Genes in the Transcriptome of P. verdolaga
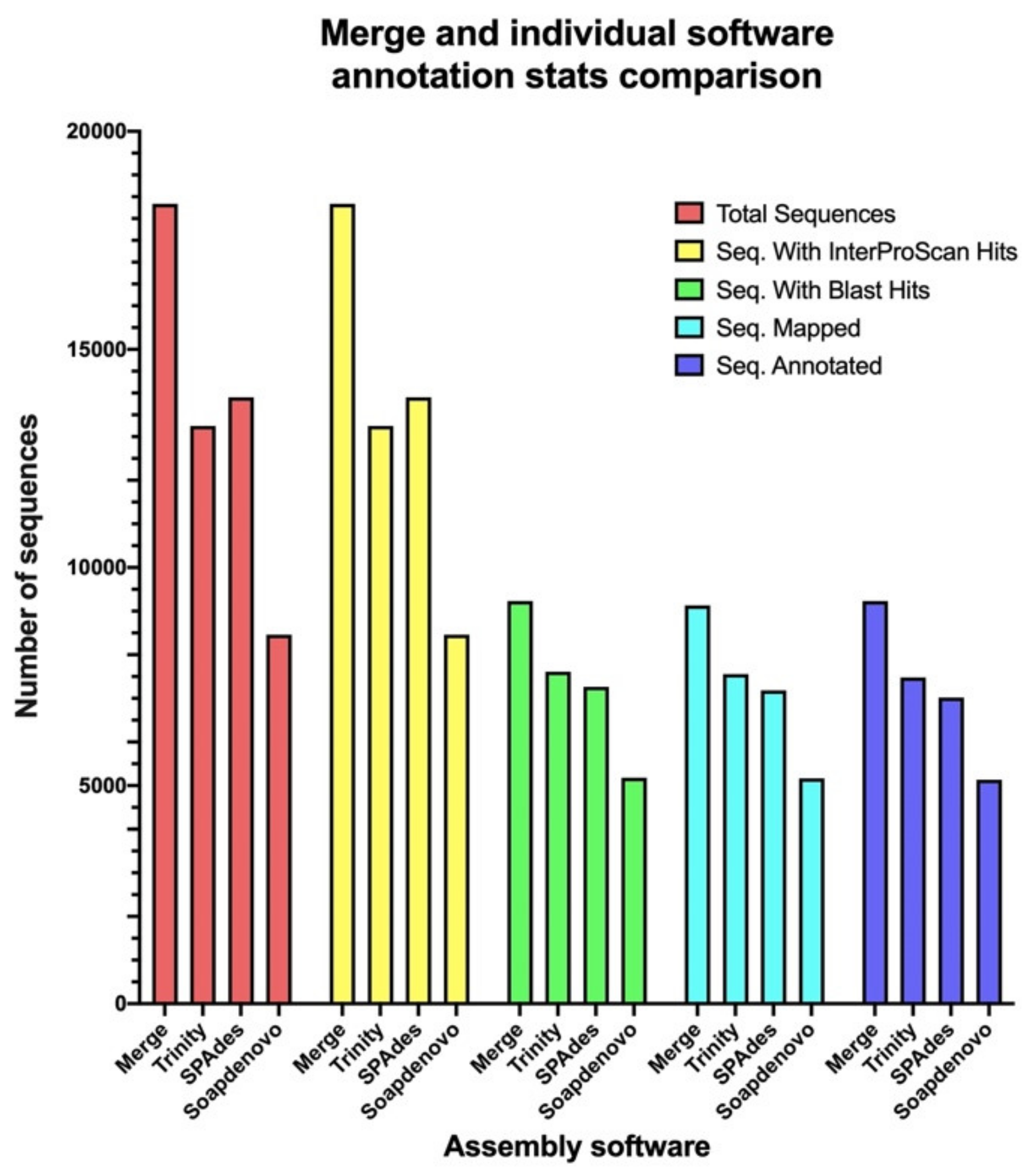
2.3. Merging of the Common and Unique Assembled ORFs for Each Software Increases the Annotation Performance
2.4. The P. verdolaga Transcriptome Is Rich in Proteins of Biotechnological Interest
3. Discussion
3.1. The Quality of the Assemblies
3.2. Structural and Functional Annotation and the Differences Observed between the Three Software
3.3. New Toxins in the Venom Gland Transcriptome of P. verdolaga
4. Conclusions
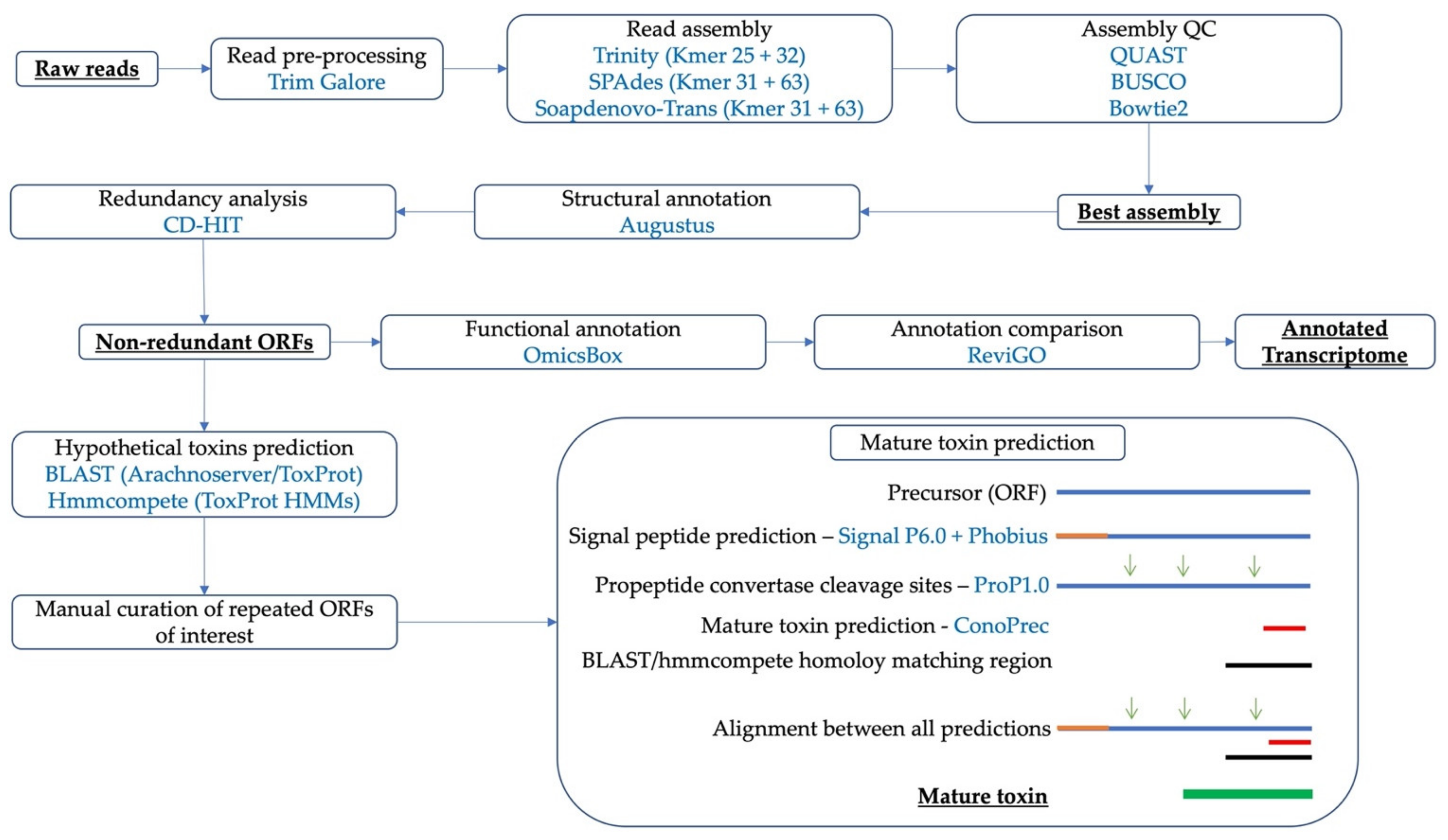
5. Materials and Methods
5.1. Venom Gland Transcriptome Data
5.2. Transcriptome Assembly and Statistics
5.3. Assessing Transcriptome Completion with BUSCO
5.4. Structural and Functional Annotation of the Transcriptome Assemblies
5.5. Prediction of Proteins and Enzymes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Spider Catalog. World Spider Catalog. Version 23.0. Natural History Museum Bern. 2022. Available online: http://wsc.nmbe.ch (accessed on 31 January 2022). [CrossRef]
- Dubovskii, P.V.; Vassilevski, A.A.; Kozlov, S.A.; Feofanov, A.V.; Grishin, E.V.; Efremov, R.G. Latarcins: Versatile spider venom peptides. Cell. Mol. Life Sci. 2015, 72, 4501–4522. [Google Scholar] [CrossRef]
- King, G.F.; Hardy, M.C. Spider-venom peptides: Structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 2013, 58, 475–496. [Google Scholar] [CrossRef] [PubMed]
- Vassilevski, A.A.; Kozlov, S.A.; Grishin, E.V. Molecular diversity of spider venom. Biochemistry 2009, 74, 1505–1534. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Modulation of insect Cav channels by peptidic spider toxins. Toxicon 2007, 49, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.G.; Dobroff, A.S.S.; Cavarsan, C.F.; Paschoalin, T.; Nimrichter, L.; Mortara, R.A.; Wu, W.; Luo, Q. Effective topical treatment of subcutaneous murine B16F10-Nex2 melanoma by the antimicrobial peptide gomesin. Neoplasia 2008, 10, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santibáñez-López, C.E.; Possani, L.D. Overview of the Knottin scorpion toxin-like peptides in scorpion venoms: Insights on their classification and evolution. Toxicon 2015, 107, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Chellat, M.F.; Raguž, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chem.-Int. Ed. 2016, 55, 6600–6626. [Google Scholar] [CrossRef]
- Yewale, V.N. Antimicrobial Resistance—A Ticking Bomb! 1st ed.; Cadman, H., Martinez, L., Eds.; Indian Pediatrics, World Health Organization: Geneva, Switzerland, 2014; Volume 51, pp. 171–172. [Google Scholar]
- Windley, M.J.; Herzig, V.; Dziemborowicz, S.A.; Hardy, M.C.; King, G.F.; Nicholson, G.M. Spider-venom peptides as bioinsecticides. Toxins 2012, 4, 191–227. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Wang, M.; Wu, W.; Luo, Q.; Jiang, L.; Tao, H.; Reis, P.V.; Pimenta, A.M.C. Spider venom peptides as potential drug candidates due to their anticancer and antinociceptive activities. J. Venom. Anim. Toxins Incl. Trop. Dis. 2019, 25, 1–13. [Google Scholar] [CrossRef]
- Saez, N.J.; Herzig, V. Versatile spider venom peptides and their medical and agricultural applications. Toxicon 2019, 158, 109–126. [Google Scholar] [CrossRef]
- Rapôso, C. Scorpion and spider venoms in cancer treatment: State of the art, challenges, and perspectives. J. Clin. Transl. Res. 2017, 3, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.D.; Deplazes, E.; Mancera, R.L. The biological and biophysical properties of the spider peptide gomesin. Molecules 2018, 23, 1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bal, V.H.; Lord, C. Replication of standardized ADOS domain scores in the Simons Simplex Collection. Autism Res. 2015, 8, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Santos, D.M.; Reis, P.V.; Pimenta, A.M.C. Spider Venoms. Spider Venoms 2015, 3, 1–15. [Google Scholar]
- Pineda, S.S.; Chaumeil, P.A.; Kunert, A.; Kaas, Q.; Thang, M.W.C.; Le, L.; Gunduz, I.; Hayes, A.; Waack, S. ArachnoServer 3.0: An online resource for automated discovery, analysis and annotation of spider toxins. Bioinformatics 2018, 34, 1074–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada-Gomez, S.; Vargas Muñoz, L.J.; Quintana Castillo, J.C. Extraction and partial characterization of venom from the Colombian spider Pamphobeteus aff. nigricolor (Aranae: Theraphosidae). Toxicon 2013, 76, 301–309. [Google Scholar] [CrossRef]
- Oldrati, V.; Koua, D.; Allard, P.M.; Hulo, N.; Arrell, M.; Nentwig, W.; Vassilevski, A.A.; Kozlov, S.A. Peptidomic and transcriptomic profiling of four distinct spider venoms. PLoS ONE 2017, 12, e0172966. [Google Scholar] [CrossRef]
- Luna-Ramírez, K.; Quintero-Hernández, V.; Juárez-González, V.R.; Possani, L.D. Whole transcriptome of the venom gland from urodacus yaschenkoi scorpion. PLoS ONE 2015, 10, e0127883. [Google Scholar] [CrossRef]
- Bouzid, W.; Verdenaud, M.; Klopp, C.; Ducancel, F.; Noirot, C.; Vétillard, A. De Novo sequencing and transcriptome analysis for tetramorium bicarinatum: A comprehensive venom gland transcriptome analysis from an ant species. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Chetia, H.; Kabiraj, D.; Singh, D.; Mosahari, P.V.; Das, S.; Sharma, P.; Johansen, A.R.; Gíslason, M.H. De novo transcriptome of the muga silkworm, Antheraea assamensis (Helfer). Gene 2017, 611, 54–65. [Google Scholar] [CrossRef]
- Gupta, S.K.; Kupper, M.; Ratzka, C.; Feldhaar, H.; Vilcinskas, A.; Gross, R.; Yun, H.-Y.; Mains, R.E. Scrutinizing the immune defence inventory of Camponotus floridanus applying total transcriptome sequencing. BMC Genom. 2015, 16, 540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, D.W. Using hidden Markov models to align multiple sequences. Cold Spring Harb. Protoc. 2009, 4, 1882. [Google Scholar] [CrossRef] [PubMed]
- Madera, M.; Gough, J. A comparison of profile hidden Markov model procedures for remote homology detection. Nucleic Acids Res. 2002, 30, 4321–4328. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Oda-Ueda, N.; Hisata, K.; Nakamura, H.; Chijiwa, T.; Hattori, S.; Reis, P.V.; Pimenta, A.M.C. Alternative mRNA splicing in three venom families underlying a possible production of divergent venom proteins of the habu snake, protobothrops flavoviridis. Toxins 2019, 11, 1192. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.C.; Luo, F.; Li, W.X. Characterization of a novel cDNA encoding a short venom peptide derived from venom gland of scorpion Buthus martensii Karsch: Trans-splicing may play an important role in the diversification of scorpion venom peptides. Peptides 2006, 27, 675–681. [Google Scholar] [CrossRef]
- Wong, E.S.W.; Belov, K. Venom evolution through gene duplications. Gene 2012, 496, 1–7. [Google Scholar] [CrossRef]
- Holding, M.L.; Margres, M.J.; Mason, A.J.; Parkinson, C.L.; Rokyta, D.R. Evaluating the performance of de novo assembly methods for venom-gland transcriptomics. Toxins 2018, 10, 249. [Google Scholar] [CrossRef] [Green Version]
- Hölzer, M.; Marz, M. De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-Seq assemblers. Gigascience 2019, 8, giz039. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Gómez, S.; Vargas-Muñoz, L.J.; Saldarriaga-Córdoba, M.; Cifuentes, Y.; Perafan, C. Identifying different transcribed proteins in the newly described Theraphosidae Pamphobeteus verdolaga. Toxicon 2017, 129, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Gomez, S.; Cardoso, F.C.; Vargas-Muñoz, L.J.; Quintana-Castillo, J.C.; Gómez, C.M.A.; Pineda, S.S.; Gunduz, I.; Hayes, A.; Waack, S. Venomic, transcriptomic, and bioactivity analyses of Pamphobeteus verdolaga venom reveal complex disulfide-rich peptides that modulate calcium channels. Toxins 2019, 11, 21. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Gómez, S.; Vargas-Muñoz, L.J.; Latorre, C.S.; Saldarriaga-Cordoba, M.M.; Arenas-Gómez, C.M. Analysis of high molecular mass compounds from the spider Pamphobeteus verdolaga venom gland. A transcriptomic and ms id approach. Toxins 2021, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.; Gera, P.; Rivera, C.; Santos, J.C. Pincho: A modular approach to high quality de novo transcriptomics. Genes 2021, 12, 2972. [Google Scholar] [CrossRef] [PubMed]
- Koua, D.; Kuhn-Nentwig, L. Spider neurotoxins, short linear cationic peptides and venom protein classification improved by an automated competition between exhaustive profile HMM classifiers. Toxins 2017, 9, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Bende, N.S.; Dziemborowicz, S.; Herzig, V.; Ramanujam, V.; Brown, G.W.; Bosmans, F.; Vassilevski, A.A.; Kozlov, S.A. The insecticidal spider toxin SFI1 is a knottin peptide that blocks the pore of insect voltage-gated sodium channels via a large β-hairpin loop. FEBS J. 2015, 282, 904–920. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.B.; Zadlock, F.J.; Zhang, Z.; Murphy, W.R.; Bentivegna, C.S. Comparison of de Novo transcriptome assemblers and k-mer strategies using the killifish, Fundulus heteroclitus. PLoS ONE 2016, 11, e0153104. [Google Scholar] [CrossRef]
- Freedman, A.H.; Clamp, M.; Sackton, T.B. Error, noise and bias in de novo transcriptome assemblies. Mol. Ecol. Resour. 2021, 21, 18–29. [Google Scholar] [CrossRef]
- Cabau, C.; Escudié, F.; Djari, A.; Guiguen, Y.; Bobe, J.; Klopp, C. Compacting and correcting Trinity and Oases RNA-Seq de novo assemblies. PeerJ 2017, 2017, e2988. [Google Scholar] [CrossRef] [Green Version]
- Sadat-Hosseini, M.; Bakhtiarizadeh, M.R.; Boroomand, N.; Tohidfar, M.; Vahdati, K. Combining independent de novo assemblies to optimize leaf transcriptome of Persian walnut. PLoS ONE 2020, 15, e0232005. [Google Scholar] [CrossRef]
- Garrison, N.L.; Rodriguez, J.; Agnarsson, I.; Coddington, J.A.; Griswold, C.E.; Hamilton, C.A.; Gunduz, I.; Hayes, A.; Waack, S. Spider phylogenomics: Untangling the Spider Tree of Life. PeerJ 2016, 2016, 1092762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Mahmood, K.; Orabi, J.; Kristensen, P.S.; Sarup, P.; Jørgensen, L.N.; Jahoor, A. De novo transcriptome assembly, functional annotation, and expression profiling of rye (Secale cereale L.) hybrids inoculated with ergot (Claviceps purpurea). Sci. Rep. 2020, 10, 13475. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.; Kallal, R.J.; Dimitrov, D.; Ballesteros, J.A.; Arnedo, M.A.; Giribet, G.; Reis, P.V.; Pimenta, A.M.C. Phylogenomics, Diversification Dynamics, and Comparative Transcriptomics across the Spider Tree of Life. Curr. Biol. 2018, 28, 1489–1497.e5. [Google Scholar] [CrossRef] [Green Version]
- Oda, H.; Akiyama-Oda, Y. The common house spider Parasteatoda tepidariorum. Evodevo 2020, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]
- NIH. NCBI Refseq. Available online: https://www.ncbi.nlm.nih.gov/refseq/ (accessed on 11 June 2020).
- Carlson, D.E.; Hedin, M. Comparative transcriptomics of Entelegyne spiders (Araneae, Entelegynae), with emphasis on molecular evolution of orphan genes. PLoS ONE 2017, 12, e019923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, S.; Lüddecke, T.; Cheng, D.Q.; Krehenwinkel, H.; Künzel, S.; Longhorn, S.J.; Johansen, A.R.; Gíslason, M.H. Tarantula phylogenomics: A robust phylogeny of deep theraphosid clades inferred from transcriptome data sheds light on the prickly issue of urticating setae evolution. Mol. Phylogenet. Evol. 2019, 140, 106573. [Google Scholar] [CrossRef]
- Cheng, T.C.; Long, R.W.; Wu, Y.Q.; Guo, Y.B.; Liu, D.L.; Peng, L.; Wu, W.; Luo, Q. Identification and characterization of toxins in the venom gland of the Chinese bird spider, Haplopelma hainanum, by transcriptomic analysis. Insect Sci. 2016, 23, 487–499. [Google Scholar] [CrossRef]
- Haney, R.A.; Ayoub, N.A.; Clarke, T.H.; Hayashi, C.Y.; Garb, J.E. Dramatic expansion of the black widow toxin arsenal uncovered by multi-tissue transcriptomics and venom proteomics. BMC Genom. 2014, 15, 366. [Google Scholar] [CrossRef] [Green Version]
- Gremski, L.H.; Da Silveira, R.B.; Chaim, O.M.; Probst, C.M.A.; Ferrer, V.P.; Nowatzki, J.; Yun, H.-Y.; Mains, R.E. A novel expression profile of the Loxosceles intermedia spider venomous gland revealed by transcriptome analysis. Mol. Biosyst. 2010, 6, 2403–2416. [Google Scholar] [CrossRef]
- He, B.; Zhao, S.; Chen, Y.; Cao, Q.; Wei, C.; Cheng, X.; Vassilevski, A.A.; Kozlov, S.A. Optimal assembly strategies of transcriptome related to ploidies of eukaryotic organisms. BMC Genom. 2015, 16, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.S.W.; Hardy, M.C.; Wood, D.; Bailey, T.; King, G.F. SVM-Based Prediction of Propeptide Cleavage Sites in Spider Toxins Identifies Toxin Innovation in an Australian Tarantula. PLoS ONE 2013, 8, e0192341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpabai Rajesh, R.; Franklin, J.B. Identification of Conotoxins with Novel Odd Number of Cysteine Residues from the Venom of a Marine Predatory Gastropod Conus leopardus Found in Andaman Sea. Protein Pept. Lett. 2018, 22, 25. [Google Scholar]
- Haakenson, J.K.; Deiss, T.C.; Warner, G.F.; Mwangi, W.; Criscitiello, M.F.; Smider, V.V. A Broad Role for Cysteines in Bovine Antibody Diversity. ImmunoHorizons 2019, 3, 478–487. [Google Scholar] [CrossRef]
- Iwaoka, M.; Mitsuji, T.; Shinozaki, R. Oxidative folding pathways of bovine milk β-lactoglobulin with odd cysteine residues. FEBS Open Bio 2019, 9, 1379–1391. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 10, 4633–4679. [Google Scholar] [CrossRef]
- Kuhn-Nentwig, L.; Langenegger, N.; Heller, M.; Koua, D.; Nentwig, W. The dual prey-inactivation strategy of spiders—In-depth venomic analysis of Cupiennius salei. Toxins 2019, 11, 299. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, S.A.; Vassilevski, A.A.; Feofanov, A.V.; Surovoy, A.Y.; Karpunin, D.V.; Grishin, E.V. Latarcins, antimicrobial and cytolytic peptides from the venom of the spider Lachesana tarabaevi (Zodariidae) that exemplify biomolecular diversity. J. Biol. Chem. 2006, 281, 20983–20992. [Google Scholar] [CrossRef] [Green Version]
- Kuhn-Nentwig, L.; Müller, J.; Schaller, J.; Walz, A.; Dathe, M.; Nentwig, W. Cupiennin 1, a new family of highly basic antimicrobial peptides in the venom of the spider Cupiennius salei (Ctenidae). J. Biol. Chem. 2002, 277, 11208–11216. [Google Scholar] [CrossRef] [Green Version]
- Pukala, T.L.; Doyle, J.R.; Llewellyn, L.E.; Kuhn-Nentwig, L.; Apponyi, M.A.; Separovic, F.; Johansen, A.R.; Gíslason, M.H. Cupiennin 1a, an antimicrobial peptide from the venom of the neotropical wandering spider Cupiennius salei, also inhibits the formation of nitric oxide by neuronal nitric oxide synthase. FEBS J. 2007, 274, 1778–1784. [Google Scholar] [CrossRef] [PubMed]
- Grolleau, F.; Stankiewicz, M.; Birinyi-Strachan, L.; Wang, X.H.; Nicholson, G.M.; Pelhate, M.; Gunduz, I.; Hayes, A.; Waack, S. Electrophysiological analysis of the neurotoxic action of a funnel-web spider toxin, δ-atracotoxin-Hv1a, on insect voltage-gated Na+ channels. J. Exp. Biol. 2001, 204, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Bloomquist, J.R. Mode of action of atracotoxin at central and peripheral synapses of insects. Invertebr. Neurosci. 2003, 5, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Rohou, A.; Nield, J.; Ushkaryov, Y.A. Insecticidal toxins from black widow spider venom. Toxicon 2007, 49, 531–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, A.C.; Rahman, M.A.; Volynski, K.E.; Manser, C.; Orlova, E.V.; Matsushita, H.; Wu, W.; Luo, Q. Tetramerisation of α-latrotoxin by divalent cations is responsible for toxin-induced non-vesicular release and contributes to the Ca2+-dependent vesicular exocytosis from synaptosomes. Biochimie 2000, 82, 453–468. [Google Scholar] [CrossRef]
- Südhof, T.C.; Starke, K. Preface. Handb. Exp. Pharmacol. 2008, 184, 1–33. [Google Scholar]
- Garb, J.E.; Hayashi, C.Y. Molecular evolution of α-latrotoxin, the exceptionally potent vertebrate neurotoxin in black widow spider venom. Mol. Biol. Evol. 2013, 30, 999–1014. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, S.; Iwanaga, S. Role of lectins in the innate immunity of horseshoe crab. Dev. Comp. Immunol. 1999, 23, 391–400. [Google Scholar] [CrossRef]
- Coelho, L.C.B.B.; Silva, P.M.D.S.; Lima, V.L.D.M.; Pontual, E.V.; Paiva, P.M.G.; Napoleão, T.H.; Yun, H.-Y.; Mains, R.E. Lectins, Interconnecting Proteins with Biotechnological/Pharmacological and Therapeutic Applications. Evid.-Based Complementary Altern. Med. 2017, 2017, 2257. [Google Scholar] [CrossRef]
- Chettri, D.; Boro, M.; Sarkar, L.; Verma, A.K. Lectins: Biological significance to biotechnological application. Carbohydr. Res. 2021, 506, 108367. [Google Scholar] [CrossRef]
- Eipper, B.A.; Milgram, S.L.; Jean Husten, E.; Yun, H.-Y.; Mains, R.E. Peptidylglycine α-amidating monooxygenase: A multifunctional protein with catalytic, processing, and routing domains. Protein Sci. 1993, 2, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Reis, P.V.; Pimenta, A.M.C. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Vassilevski, A.A.; Kozlov, S.A. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. RnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 2019, 8, giz100. [Google Scholar] [CrossRef] [Green Version]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33 (Suppl. S2), 465–467. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e011234. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Yun, H.-Y.; Mains, R.E. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Vassilevski, A.A.; Kozlov, S.A. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 6, 8721. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 2007, 35 (Suppl. S2), 429–432. [Google Scholar] [CrossRef] [Green Version]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Duckert, P.; Brunak, S.; Blom, N. Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 2004, 17, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trinity k-mer 25 | Trinity k-mer 32 | SPAdes k-mer 31 | SPAdes k-mer 63 | SOAPdenovo-Trans k-mer 31 | SOAPdenovo-Trans k-mer 63 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of contigs > 200 bp | 99,316 | 96,821 | 80,030 | 81,831 | 68,700 | 59,910 | ||||
| Number of contigs < 200 bp | 0 | 0 | 58,250 | 32,694 | 1,348,391 | 169,055 | ||||
| %GC | 39.5 | 39.82 | 39.42 | 39.89 | 38.93 | 40.21 | ||||
| N50 | 772 | 797 | 882 | 722 | 468 | 504 | ||||
| Number of nt | 56,851,075 | 56,627,550 | 49,124,765 | 44,839,329 | 29,425,929 | 26,911,362 | ||||
| Raw read representation in contigs | 95.68% | 96.82% | 96.94% | 94.72% | 16.66% | 92.47% | ||||
| P. tepitadorium al. average length | 14,083 | 11,069 | 11,269 | 7819 | 7091 | 5050 | ||||
| P. tepitadorium al. total coverage | 29,236,516 | 25,625,827 | 17,635,778 | 15,521,127 | 3,148,352 | 6,004,386 | ||||
| P. tepitadorium al. average quality | 14 | 12 | 15 | 11 | 20 | 11 | ||||
| P. tepitadorium al. max length | 247,989 | 247,989 | 247,989 | 247,989 | 106,194 | 168,652 | ||||
| P. tepitadorium al. min length | 40 | 40 | 40 | 40 | 40 | 40 | ||||
| Size | >200 bp | >200 bp | <200 bp | >200 bp | <200 bp | >200 bp | <200 bp | >200 bp | <200 bp | >200 bp |
| Complete BUSCOs | 79.36% | 77.95% | 0% | 79.36% | 0% | 74.01% | 0% | 46.15% | 0.1% | 58.26% |
| Fragmented BUSCOs | 9.1% | 9.47% | 3.65% | 9.01% | 3.28% | 12.76% | 16.51% | 32.18% | 6.38% | 22.42% |
| Missing BUSCOs | 11.53% | 12.57% | 96.34% | 11.63% | 96.72% | 13.23% | 83.49% | 21.67% | 93.52% | 19.32% |
| Duplicated BUSCOs | 24.23% | 23.70% | 0% | 8.98% | 0% | 6.46% | 0% | 1.42% | 0% | 2.25% |
| Number of ORFs | Average Length (aa) | Maximum Length (aa) | Total Amino Acid Number | % Redundant ORFs | Number of nr ORFs | Number of Uniquely Assembled ORFs | |
|---|---|---|---|---|---|---|---|
| Trinity k-mer 25 | 17,706 | 287.5 | 4077 | 5,089,960 | 25.20 | 13,244 | 5112 |
| SPAdes k-mer 31 | 15,858 | 239.3 | 3777 | 3,795,493 | 12.31 | 13,905 | 5085 |
| SOAPdenovo-Trans k-mer 63 | 8465 | 217.7 | 1790 | 1,842,643 | 0.07 | 8459 | 942 |
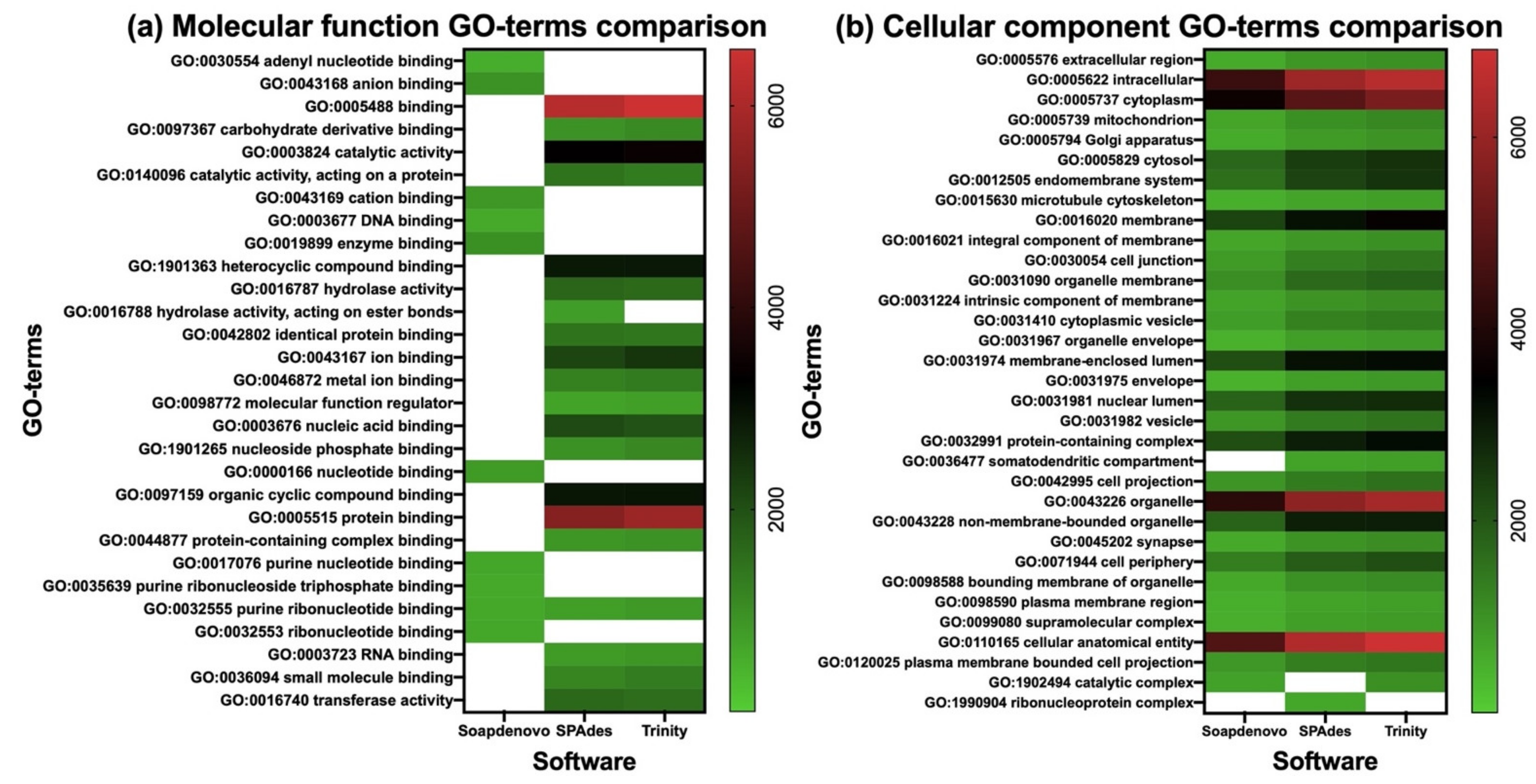
| GO-Term | SOAPdenovo-Trans | SPAdes | Trinity | ||||
|---|---|---|---|---|---|---|---|
| Category | Total Terms | Identified Terms | Associated Genes | Identified Terms | Associated Genes | Identified Terms | Associated Genes |
| Molecular function | 29 | 10 | 7086 | 20 | 39,266 | 19 | 39,986 |
| Cellular component | 33 | 31 | 45,010 | 32 | 64,844 | 32 | 71,026 |
| Biological process | 113 | 93 | 122,853 | 102 | 190,438 | 100 | 202,291 |
| Merged File | Common ORFs | Unique Trinity ORFs | Unique SPAdes ORFs | Unique SOAPdenovo-Trans ORFs | |
|---|---|---|---|---|---|
| Total sequences | 18,338 | 7199 | 5112 | 5085 | 942 |
| Annotated | 9232 | 4552 | 2703 | 1695 | 282 |
| Without annotation | 9106 | 2647 | 2409 | 3390 | 660 |
| Contribution to sequences without annotation | NA | 29.07% | 26.45% | 37.23% | 7.25% |
| GO-Term | Merge | ||||
|---|---|---|---|---|---|
| Category | Total Terms | Identified Terms | Associated Genes | Unique Terms | Missing Terms |
| Molecular function | 29 | 19 | 51,542 | - | “GO:0098772 molecular function regulator” |
| - | “GO:0032555 purine ribonucleotide binding” | ||||
| Cellular component | 33 | 31 | 69,249 | - | “GO:0110165 cellular anatomical entity” |
| Biological process | 118 | 97 | 216,509 | - | “GO:0003008 system process” |
| - | “GO:0006955 immune response” | ||||
| “GO:1901698 response to nitrogen compound” | “GO:0009410 response to xenobiotic stimulus” | ||||
| - | “GO:0015833 peptide transport” | ||||
| - | “GO:0018193 peptidyl-amino acid modification” | ||||
| “GO:0051049 regulation of transport” | “GO:0032880 regulation of protein localization” | ||||
| - | “GO:0032940 secretion by cell” | ||||
| “GO:0015031 protein transport” | “GO:0033365 protein localization to organelle” | ||||
| “GO:2000026 reg. of multicell. orga. develop.” | “GO:0045595 regulation of cell differentiation” | ||||
| - | “GO:0046903 secretion” | ||||
| - | “GO:0140352 export from cell” | ||||
| “GO:0044770 cell cycle phase transition” | “GO:1903047 mitotic cell cycle process” | ||||
| ArachnoServer | ToxProt | |||||||
|---|---|---|---|---|---|---|---|---|
| Associated Keyword | Trinity | SPAdes | SOAPdenovo-Trans | Merge | Trinity | SPAdes | SOAPdenovo-Trans | Merge |
| Antimicrobial | 0 | 4 | 0 | 4 | 5 | 8 | 2 | 9 |
| Antinociceptive | 1 | 1 | 0 | 1 | 1 | 4 | 0 | 5 |
| Antiparasitic | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Antiarrhythmic | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Cytolytic | 2 | 2 | 2 | 2 | 0 | 0 | 0 | 0 |
| Hemolytic | 2 | 2 | 2 | 2 | 3 | 8 | 2 | 8 |
| Hyaluronidase | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Pro-inflammatory | 1 | 1 | 0 | 1 | 2 | 2 | 4 | 2 |
| Kinin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Lectin | 17 | 22 | 8 | 24 | 0 | 4 | 0 | 4 |
| Necrotic | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Neurotoxin | 19 | 22 | 4 | 26 | 89 | 78 | 47 | 108 |
| Neurotransmitter hydrolysis | 3 | 4 | 1 | 4 | 0 | 0 | 0 | 0 |
| Presynaptic neurotoxin | 66 | 50 | 41 | 70 | 64 | 38 | 39 | 69 |
| Protease inhibitor | 13 | 9 | 4 | 14 | 12 | 9 | 4 | 13 |
| Protease | 22 | 18 | 15 | 22 | 0 | 0 | 0 | 0 |
| Total | 150 | 139 | 79 | 174 | 178 | 153 | 100 | 220 |
| hmmcompete’s HMMs | Trinity | SPAdes | SOAPdenovo-Trans | Merge |
|---|---|---|---|---|
| Cationic peptides | 7 | 9 | 6 | 8 |
| Neurotoxins | 58 | 53 | 24 | 77 |
| Venom proteins | 221 | 180 | 132 | 233 |
| Total | 286 | 242 | 162 | 318 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salinas-Restrepo, C.; Misas, E.; Estrada-Gómez, S.; Quintana-Castillo, J.C.; Guzman, F.; Calderón, J.C.; Giraldo, M.A.; Segura, C. Improving the Annotation of the Venom Gland Transcriptome of Pamphobeteus verdolaga, Prospecting Novel Bioactive Peptides. Toxins 2022, 14, 408. https://doi.org/10.3390/toxins14060408
Salinas-Restrepo C, Misas E, Estrada-Gómez S, Quintana-Castillo JC, Guzman F, Calderón JC, Giraldo MA, Segura C. Improving the Annotation of the Venom Gland Transcriptome of Pamphobeteus verdolaga, Prospecting Novel Bioactive Peptides. Toxins. 2022; 14(6):408. https://doi.org/10.3390/toxins14060408
Chicago/Turabian StyleSalinas-Restrepo, Cristian, Elizabeth Misas, Sebastian Estrada-Gómez, Juan Carlos Quintana-Castillo, Fanny Guzman, Juan C. Calderón, Marco A. Giraldo, and Cesar Segura. 2022. "Improving the Annotation of the Venom Gland Transcriptome of Pamphobeteus verdolaga, Prospecting Novel Bioactive Peptides" Toxins 14, no. 6: 408. https://doi.org/10.3390/toxins14060408