Deoxynivalenol Degradation by Various Microbial Communities and Its Impacts on Different Bacterial Flora

 ,
, 
Abstract
:1. Introduction
2. Results
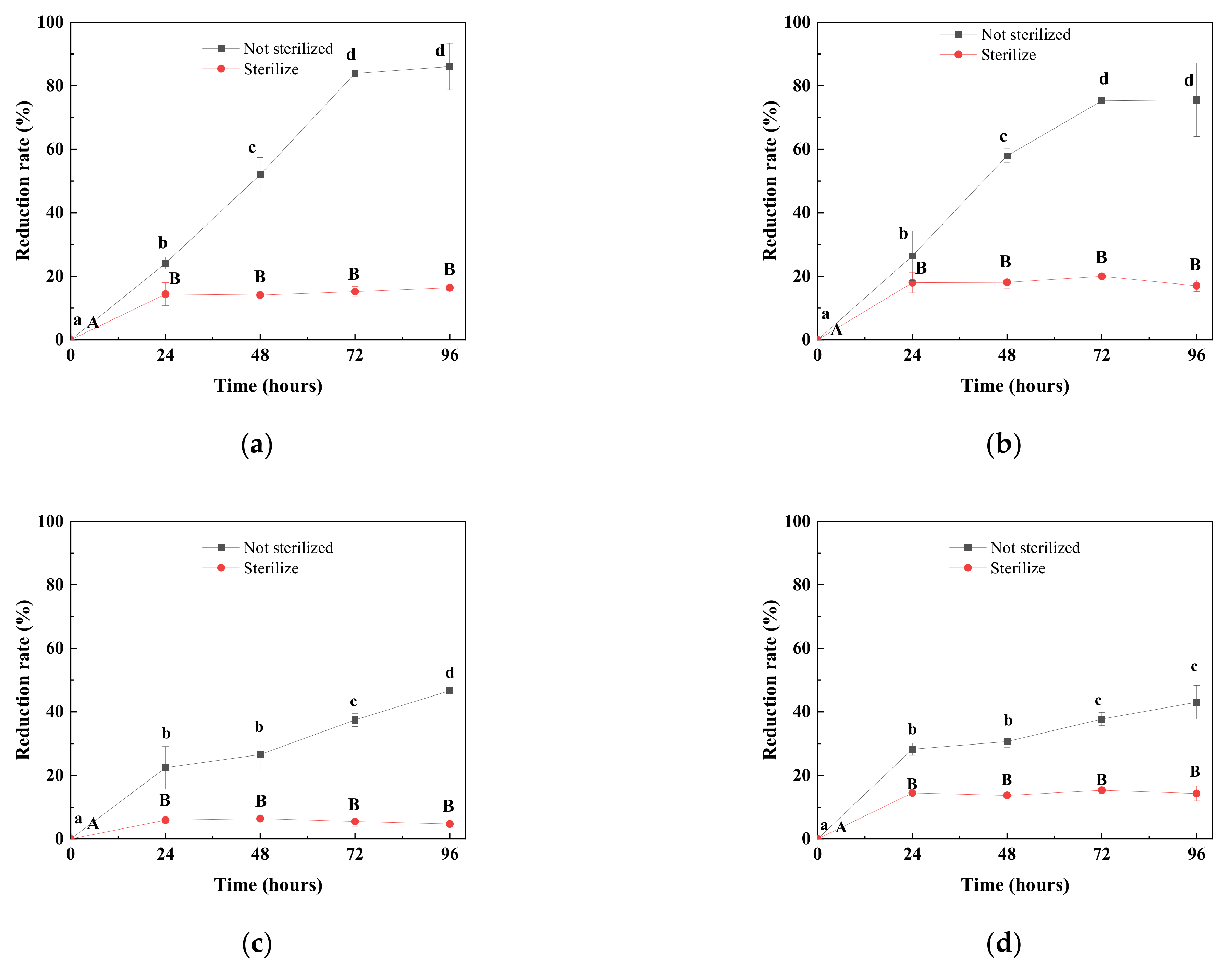
2.1. Degradation of DON by Different Natural Microbial Communities
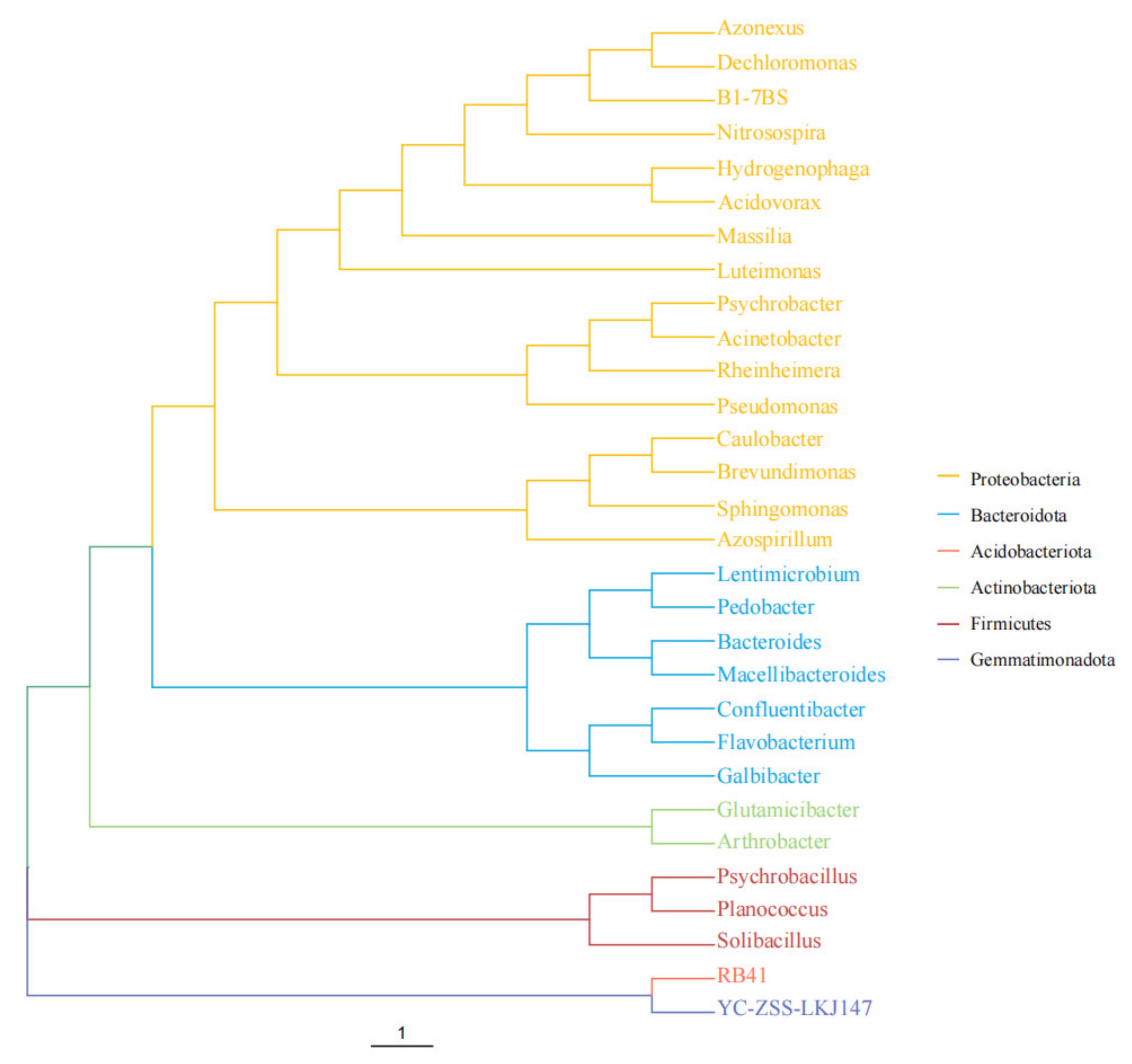
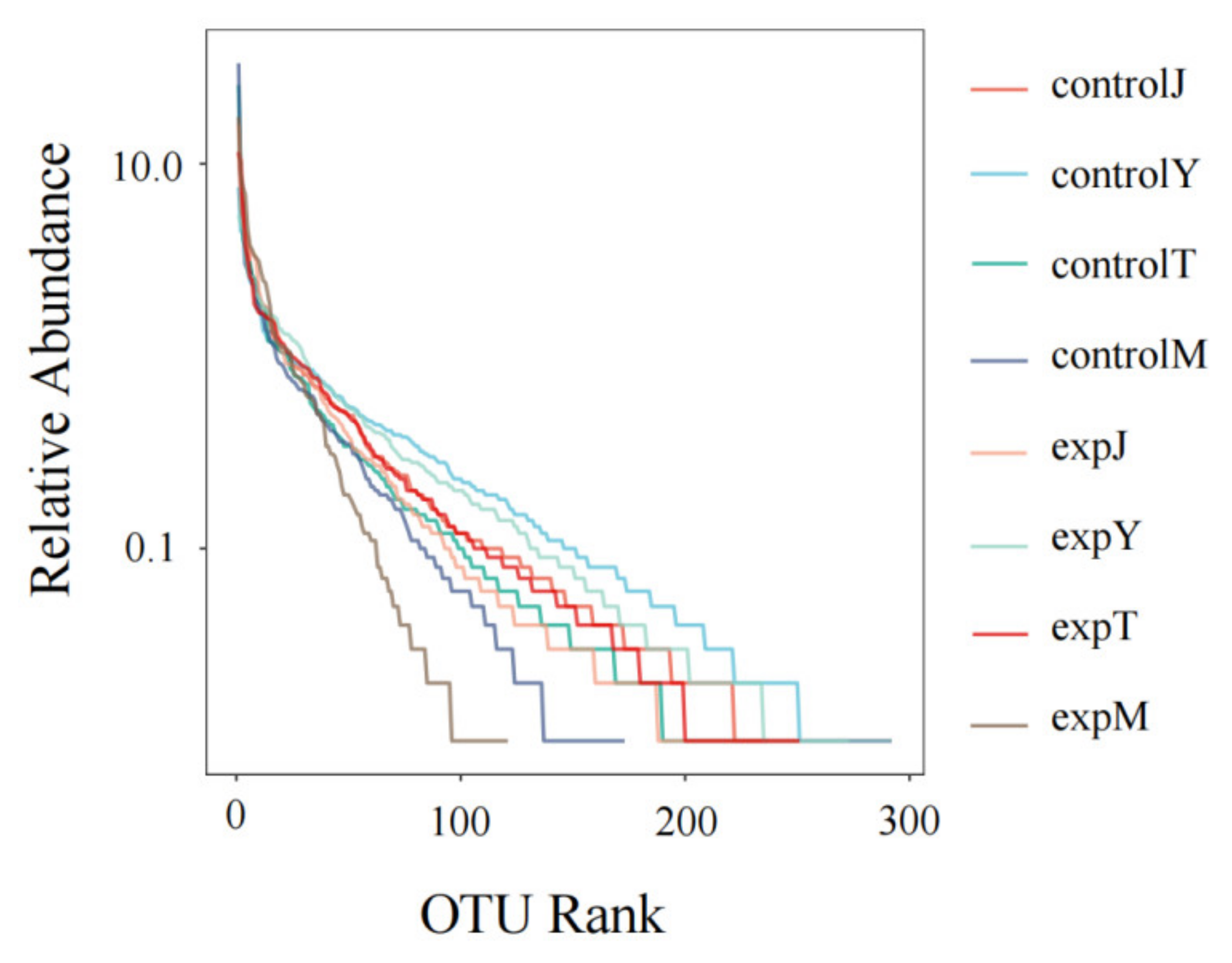
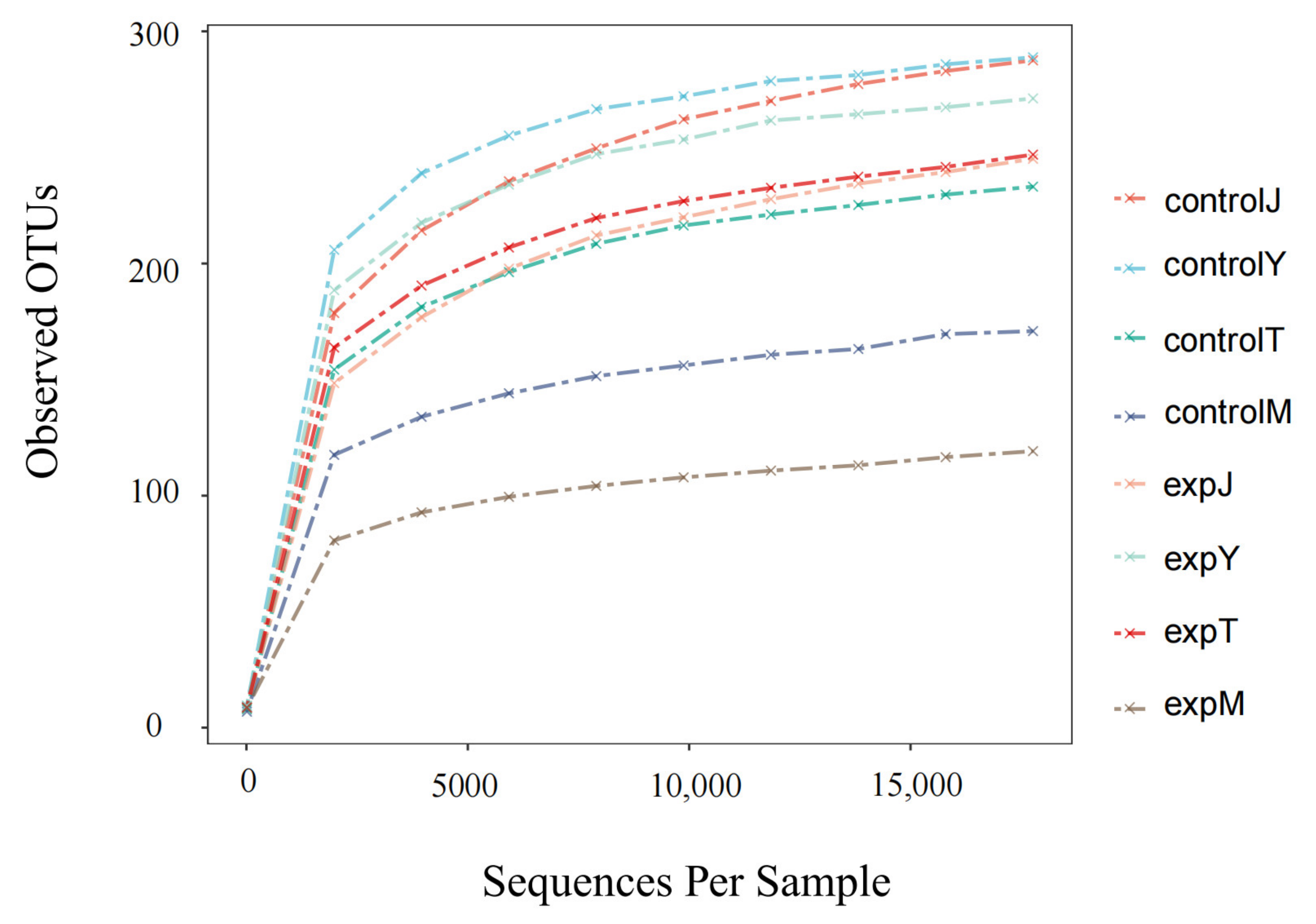
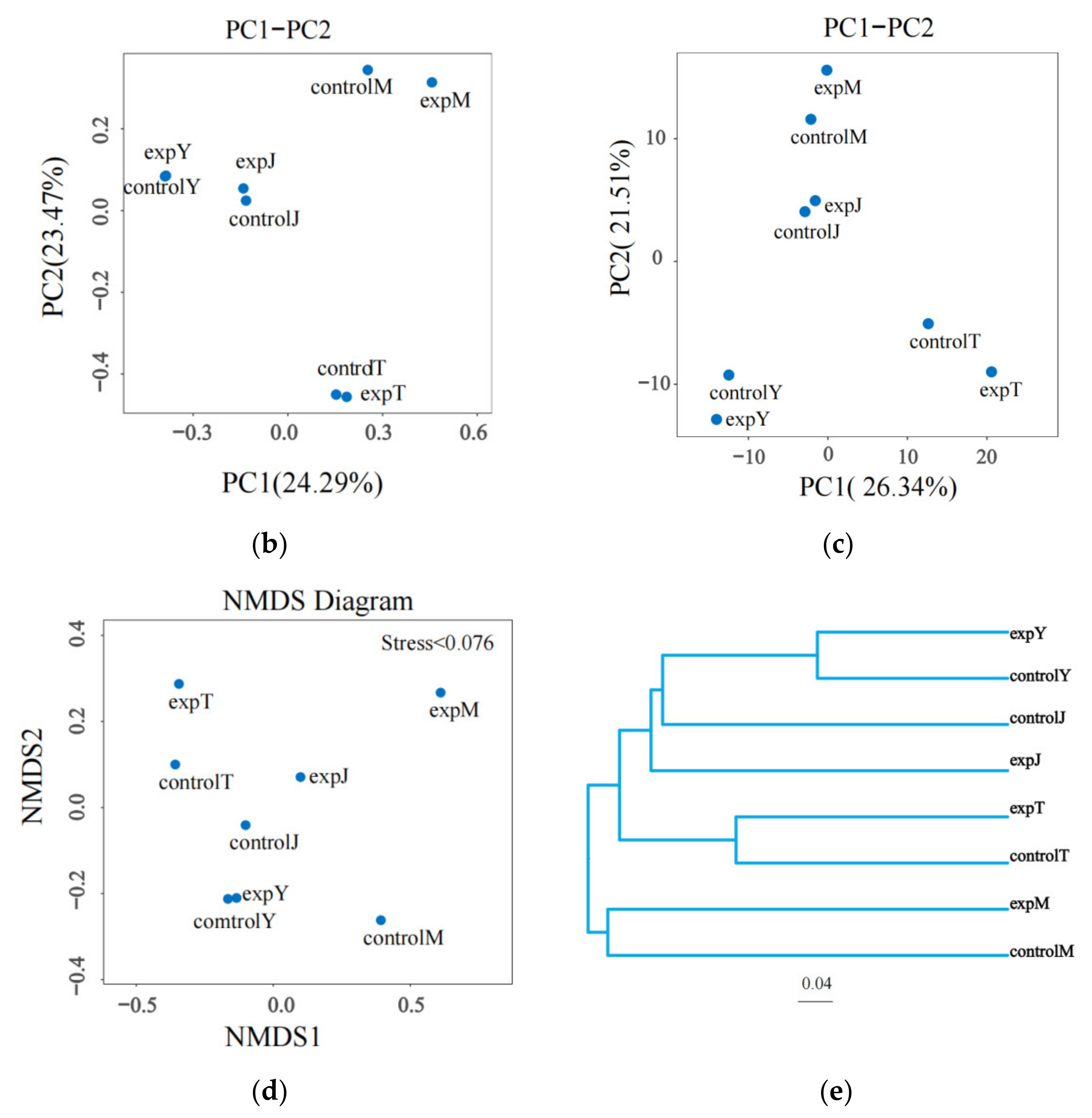
2.2. The Diversities of Bacterial Flora in the DON-Degrading Samples
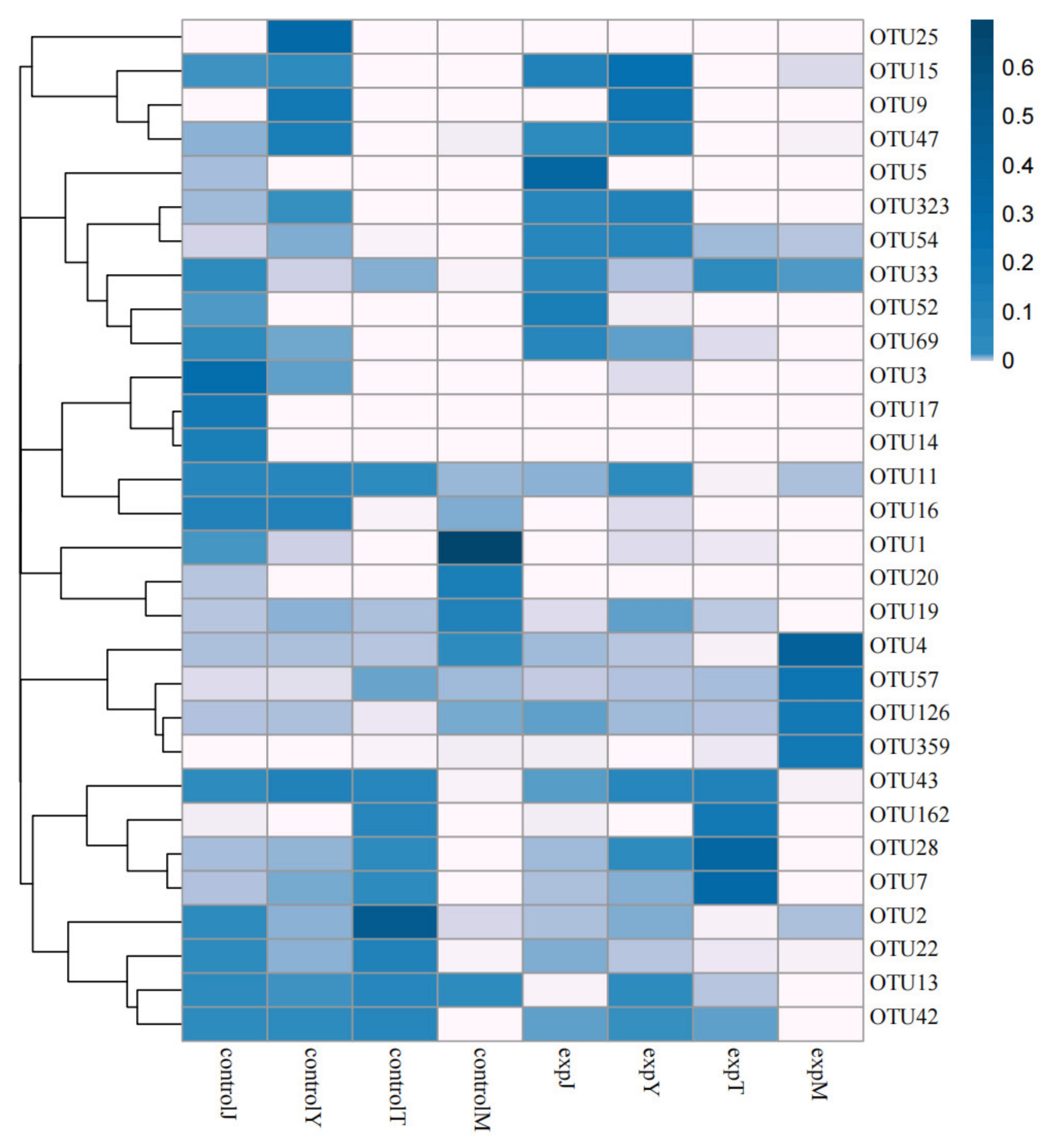
2.2.1. OTU Cluster Analysis
2.2.2. Alpha Diversity Analysis
2.2.3. Beta Diversity Analysis
3. Discussion
4. Materials and Methods
4.1. Samples and Chemicals
4.2. DON Degradation by Microbial Communities
4.3. Microbial Diversity Analysis of Mixed Flora in Samples
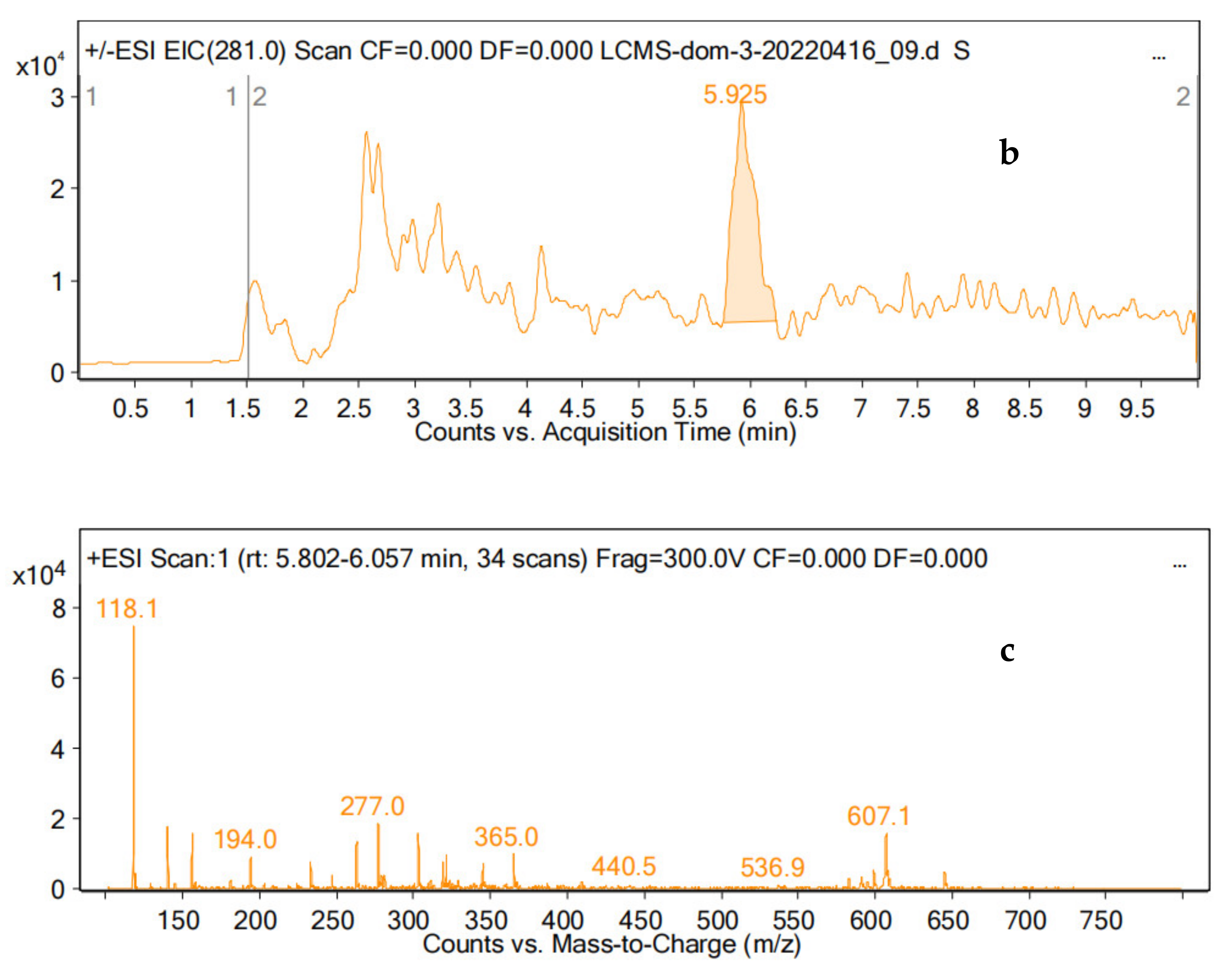
4.4. DON Analysis
4.5. Calculation of DON Degradation Rate
4.6. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bracarense, A.; Pierron, A.; Pinton, P.; Gerez, J.R.; Schatzmayr, G.; Moll, W.D.; Zhou, T.; Oswald, I.P. Reduced toxicity of 3-epi-deoxynivalenol and de-epoxy-deoxynivalenol through deoxynivalenol bacterial biotransformation: In vivo analysis in piglets. Food Chem. Toxicol. 2020, 140, 111241. [Google Scholar] [CrossRef] [PubMed]
- Audenaert, K.; Vanheule, A.; Höfte, M.; Haesaert, G. Deoxynivalenol: A Major player in the multifaceted response of Fusarium to its environment. Toxins 2013, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Magan, N.; Hope, R.; Colleate, A.; Baxter, E.S. Relationship between growth and mycotoxin production by Fusarium species, biocides and environment. Eur. J. Plant Pathol. 2002, 108, 685–690. [Google Scholar] [CrossRef]
- Nasaruddin, N.; Jinap, S.; Samsudin, N.I.P.; Kamarulzaman, N.H.; Sanny, M. Assessment of multi-mycotoxin contamination throughout the supply chain of maize-based poultry feed from selected regions of Malaysia by LC-MS/MS. Food Add. Cont. Part A Chem. Anal. Control Exp. Risk Asses. 2022, 39, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Milicevic, D.; Nastasijevic, I.; Petrovic, Z. Mycotoxin in the food supply chainimplications for public health program. J. Environ. Sci. Health Part C Environ. Carcinogen. Ecotoxicol. Rev. 2016, 34, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, H.K.; Alexander, J.; Barregard, L.; Bignami, M.; Bruschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Grasl-Kraupp, B.; Hogstrand, C. Risks to human and animal health related to the presence of deoxynivalenol and its acetylated and modified forms in food and feed. EFSA Panel Contam. Food Chain. 2017, 15, 4718. [Google Scholar]
- Dai, Y.J.; Xie, H.Q.; Xu, Y.X. Evaluation of deoxynivalenol-induced toxic effects on mouse endometrial stromal cells: Cell apoptosis and cell cycle. Biochem. Biophys. Res. Commun. 2017, 483, 572–577. [Google Scholar] [CrossRef]
- Hou, S.L.; Ma, J.J.; Cheng, Y.Q.; Wang, H.A.; Sun, J.H.; Yan, Y.X. The toxicity mechanisms of DON to humans and animals and potential biological treatment strategies. Crit. Rev. Food Sci. Nutr. 2021, 9, 1–23. [Google Scholar] [CrossRef]
- Mishra, S.; Srivastava, S.; Dewangan, J.; Divakar, A.; Kumar Rath, S. Global occurrence of deoxynivalenol in food commodities and exposure risk assessment in humans in the last decade: A survey. Crit. Rev. Food Sci. Nutr. 2020, 60, 1346–1374. [Google Scholar] [CrossRef]
- Ji, F.; Xu, J.; Liu, X.; Yin, X.; Shi, J. Natural occurrence of deoxynivalenol and zearalenone in wheat from jiangsu province, china. Food Chem. 2014, 157, 393–397. [Google Scholar] [CrossRef]
- Birr, T.; Jensen, T.; Preusske, N.; Sonnichsen, F.D.; De Boevre, M.; De Saeger, S.; Hasler, M.; Verreet, J.A.; Klink, H. Occurrence of fusarium mycotoxins and their modified forms in forage maize cultivars. Toxins 2021, 13, 110. [Google Scholar] [CrossRef]
- Righetti, L.; Damiani, T.; Rolli, E.; Galaverna, G.; Suman, M.; Bruni, R.; Dall’Asta, C. Exploiting the potential of micropropagated durum wheat organs as modified mycotoxin biofactories: The case of deoxynivalenol. Phytochemistry 2020, 170, 112194. [Google Scholar] [CrossRef]
- Ji, X.; Qiao, Y.; Zheng, W.; Jiang, H.; Yao, W. Deoxynivalenol interferes with intestinal motility via injuring the contractility of enteric smooth muscle cells: A novel hazard to the gastrointestinal tract by environmental toxins. Ecotoxicol. Environ. Saf. 2021, 224, 112656. [Google Scholar] [CrossRef]
- Al-Jaal, B.A.; Jaganjac, M.; Barcaru, A.; Horvatovich, P.; Latiff, A. Aflatoxin, fumonisin, ochratoxin, zearalenone and deoxynivalenol biomarkers in human biological fluids: A systematic literature review, 2001–2018. Food Chem. Toxicol. 2019, 129, 211–228. [Google Scholar] [CrossRef]
- James, J.P.; Alexa, T.S. Deoxynivalenol: Toxicology and potential effects on humans. J. Toxicol. Environ. Health Part B Crit. Rev. 2005, 8, 39–69. [Google Scholar]
- Bonnet, M.S.; Roux, J.; Mounien, L.; Dallaporta, M.; Troadec, J.D. Advances in deoxynivalenol toxicity mechanisms: The brain as target. Toxins 2012, 4, 1120–1138. [Google Scholar] [CrossRef]
- McCormick, S.P. Microbial detoxification of mycotoxins. J. Chem. Ecol. 2013, 39, 907–918. [Google Scholar] [CrossRef]
- Guo, H.; Ji, J.; Wang, J.S.; Sun, X. Deoxynivalenol: Masked forms, fate during food processing, and potential biological remedies. Compr. Rev. Food Sci. Food Safety. 2020, 19, 895–926. [Google Scholar] [CrossRef]
- Gao, X.; Mu, P.; Zhu, X.; Chen, X.; Tang, S.; Wu, Y.; Miao, X.; Wang, X.; Wen, J.; Deng, Y. Dual function of a novel bacterium, slackia sp. D-g6: Detoxifying deoxynivalenol and producing the natural estrogen analogue, equol. Toxins 2020, 12, 85. [Google Scholar] [CrossRef]
- Maidana, L.G.; Gerez, J.; Pinho, F.; Garcia, S. Bracarense, Apfl. Lactobacillus plantarum culture supernatants improve intestinal tissue exposed to deoxynivalenol. Exp. Toxicol. Pathol. 2017, 69, 666–671. [Google Scholar] [CrossRef]
- He, C.; Fan, Y.; Liu, G.; Zhang, H. Isolation and identification of a strain of aspergillus tubingensis with deoxynivalenol biotransformation capability. Int. J. Mol. Sci. 2008, 9, 2366–2375. [Google Scholar] [CrossRef] [PubMed]
- Carere, J.; Hassan, Y.I.; Lepp, D.; Zhou, T. The enzymatic detoxification of the mycotoxin deoxynivalenol: Identification of depa from the don epimerization pathway. Microb. Biotechnol. 2018, 11, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Carere, J.; Hassan, Y.I.; Lepp, D.; Zhou, T. The identification of depb: An enzyme responsible for the final detoxification step in the deoxynivalenol epimerization pathway in Devosia mutans 17-2-E-8. Front. Microbiol. 2018, 9, 1573. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Zhou, T.; Young, J.C.; Goodwin, P.H.; Pauls, K.P. Aerobic and anaerobic de-epoxydation of mycotoxin deoxynivalenol by bacteria originating from agricultural soil. World J. Microbiol. Biotechnol. 2012, 28, 7–13. [Google Scholar] [CrossRef]
- Volkl, A.; Vogler, B.; Schollenberger, M.; Karlovsky, P. Microbial detoxification of mycotoxin deoxynivalenol. J. Basic Microbiol. 2004, 44, 147–156. [Google Scholar] [CrossRef]
- Gao, H.; Niu, J.F.; Yang, H.; Lu, Z.X.; Chen, M.R.; Lv, F.X. High-throughput sequencing analysis of vomitoxin-degrading bacteria in bacterial consortium. Food Sci. 2021, 42, 123–130. [Google Scholar]
- Hassan, Y.I.; Lepp, D.; Zhou, T. Genome assemblies of three soil-associated Devosia species: D. insulae, D. limi, and D. soli. Genome Announc. 2015, 3, e00514-15. [Google Scholar] [CrossRef]
- Zhu, Y.; Hassan, Y.I.; Lepp, D.; Shao, S.; Zhou, T. Strategies and methodologies for developing microbial detoxification systems to mitigate mycotoxins. Toxins 2017, 9, 130. [Google Scholar] [CrossRef]
- Li, X.Z.; Zhu, C.; de Lange, C.F.; Zhou, T.; He, J.; Yu, H.; Gong, J.; Young, J.C. Efficacy of detoxification of deoxynivalenol-contaminated corn by Bacillus sp. LS100 in reducing the adverse effects of the mycotoxin on swine growth performance. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 894–901. [Google Scholar] [CrossRef]
- He, P.; Young, L.G.; Forsberg, C. Microbial transformation of deoxynivalenol (vomitoxin). Appl. Environ. Microbiol. 1992, 58, 3857–3863. [Google Scholar] [CrossRef]
- He, J.W.; Hassan, Y.I.; Perilla, N.; Li, X.Z.; Boland, G.J.; Zhou, T. Bacterial epimerization as a route for deoxynivalenol detoxification: The influence of growth and environmental conditions. Front. Microbiol. 2016, 7, 572. [Google Scholar] [CrossRef]
- Sato, I.; Ito, M.; Ishizaka, M.; Ikunaga, Y.; Sato, Y.; Yoshida, S.; Koitabashi, M.; Tsushima, S. Thirteen novel deoxynivalenol-degrading bacteria are classified within two genera with distinct degradation mechanisms. FEMS Microbiol. Lett. 2012, 327, 110–117. [Google Scholar] [CrossRef]
- Shima, J.; Takase, S.; Takahashi, Y.; Iwai, Y.; Fujimoto, H.; Yamazaki, M.; Ochi, K. Novel detoxification of the trichothecene mycotoxin deoxynivalenol by a soil bacterium isolated by enrichment culture. Appl. Environl. Microbiol. 1997, 63, 3825–3830. [Google Scholar] [CrossRef]
- He, J.W.; Bondy, G.S.; Zhou, T.; Caldwell, D.; Boland, G.J.; Scott, P.M. Toxicology of 3-epi-deoxynivalenol, a deoxynivalenol-transformation product by Devosia mutans 17-2-E-8. Food Chem. Toxicol. 2015, 84, 250–259. [Google Scholar] [CrossRef]
- Ikunaga, Y.; Sato, I.; Grond, S.; Numaziri, N.; Yoshida, S.; Yamaya, H.; Hiradate, S.; Hasegawa, M.; Toshima, H.; Koitabashi, M.; et al. Nocardioides sp. strain WSN05-2, isolated from a wheat field, degrades deoxynivalenol, producing the novel intermediate 3-epi-deoxynivalenol. Appl. Microbiol. Biotechnol. 2011, 89, 419–427. [Google Scholar] [CrossRef]
- Prairie Agricultural Machinery Institue (PAMI). Evaluation of Disposal Options for Fusarium-Damaged Grain and Screenings; Prairie Agricultural Machinery Institue (PAMI): Humboldt, SK, Canada, 2017; Project No. R8415. [Google Scholar]
- Du, G.A.; Liu, L.; Guo, Q.; Cui, Y.Y.; Chen, H.; Yuan, Y.H.; Wang, Z.L.; Gao, Z.P.; Sheng, Q.L.; Yue, T.L. Microbial community diversity associated with Tibetan kefir grains and its detoxification of Ochratoxin A during fermentation. Food Microbiol. 2021, 99, 103803. [Google Scholar] [CrossRef]
- Ahad, R.; Zhou, T.; Lepp, D.; Pauls, K.P. Microbial detoxification of eleven food and feed contaminating trichothecene mycotoxins. BMC Biotechnol. 2017, 17, 30. [Google Scholar] [CrossRef]
- Wilson, N.M.; McMaster, N.; Gantulga, D.; Soyars, C.; McCormick, S.P.; Knott, K.; Senger, R.S.; Schmale, D.G. Modification of the mycotoxin deoxynivalenol using microorganisms isolated from environmental samples. Toxins 2017, 9, 141. [Google Scholar] [CrossRef]
- Carter, M.R.; Gregorich, E.G. Soil Sampling and Method of Analysis, Second Edition, 2nd ed.; Taylor & Francis, Group, Ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ace | Chao1 | Shannon | Simpson | Goods_Coverage |
|---|---|---|---|---|---|
| controlJ | 320.504 | 316.375 | 6.073 | 0.967 | 0.998 |
| controlY | 305.279 | 304.65 | 6.732 | 0.982 | 0.999 |
| controlT | 256.036 | 259.895 | 5.379 | 0.92 | 0.998 |
| controlM | 197.767 | 200.083 | 4.791 | 0.875 | 0.999 |
| expJ | 277.625 | 282.913 | 5.683 | 0.955 | 0.998 |
| expY | 281.069 | 279.955 | 6.575 | 0.982 | 0.999 |
| expT | 282.15 | 297.867 | 5.969 | 0.964 | 0.998 |
| expM | 144.405 | 184.25 | 4.886 | 0.938 | 0.999 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, C.; Zhao, M.; Yao, F.; Zhu, R.; Cai, H.; Shao, S.; Li, X.-Z.; Zhou, T. Deoxynivalenol Degradation by Various Microbial Communities and Its Impacts on Different Bacterial Flora. Toxins 2022, 14, 537. https://doi.org/10.3390/toxins14080537
Cai C, Zhao M, Yao F, Zhu R, Cai H, Shao S, Li X-Z, Zhou T. Deoxynivalenol Degradation by Various Microbial Communities and Its Impacts on Different Bacterial Flora. Toxins. 2022; 14(8):537. https://doi.org/10.3390/toxins14080537
Chicago/Turabian StyleCai, Chenggang, Miaomiao Zhao, Feng Yao, Ruiyu Zhu, Haiying Cai, Suqin Shao, Xiu-Zhen Li, and Ting Zhou. 2022. "Deoxynivalenol Degradation by Various Microbial Communities and Its Impacts on Different Bacterial Flora" Toxins 14, no. 8: 537. https://doi.org/10.3390/toxins14080537
APA StyleCai, C., Zhao, M., Yao, F., Zhu, R., Cai, H., Shao, S., Li, X.-Z., & Zhou, T. (2022). Deoxynivalenol Degradation by Various Microbial Communities and Its Impacts on Different Bacterial Flora. Toxins, 14(8), 537. https://doi.org/10.3390/toxins14080537