Characterization of Lung Injury following Abrin Pulmonary Intoxication in Mice: Comparison to Ricin Poisoning

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
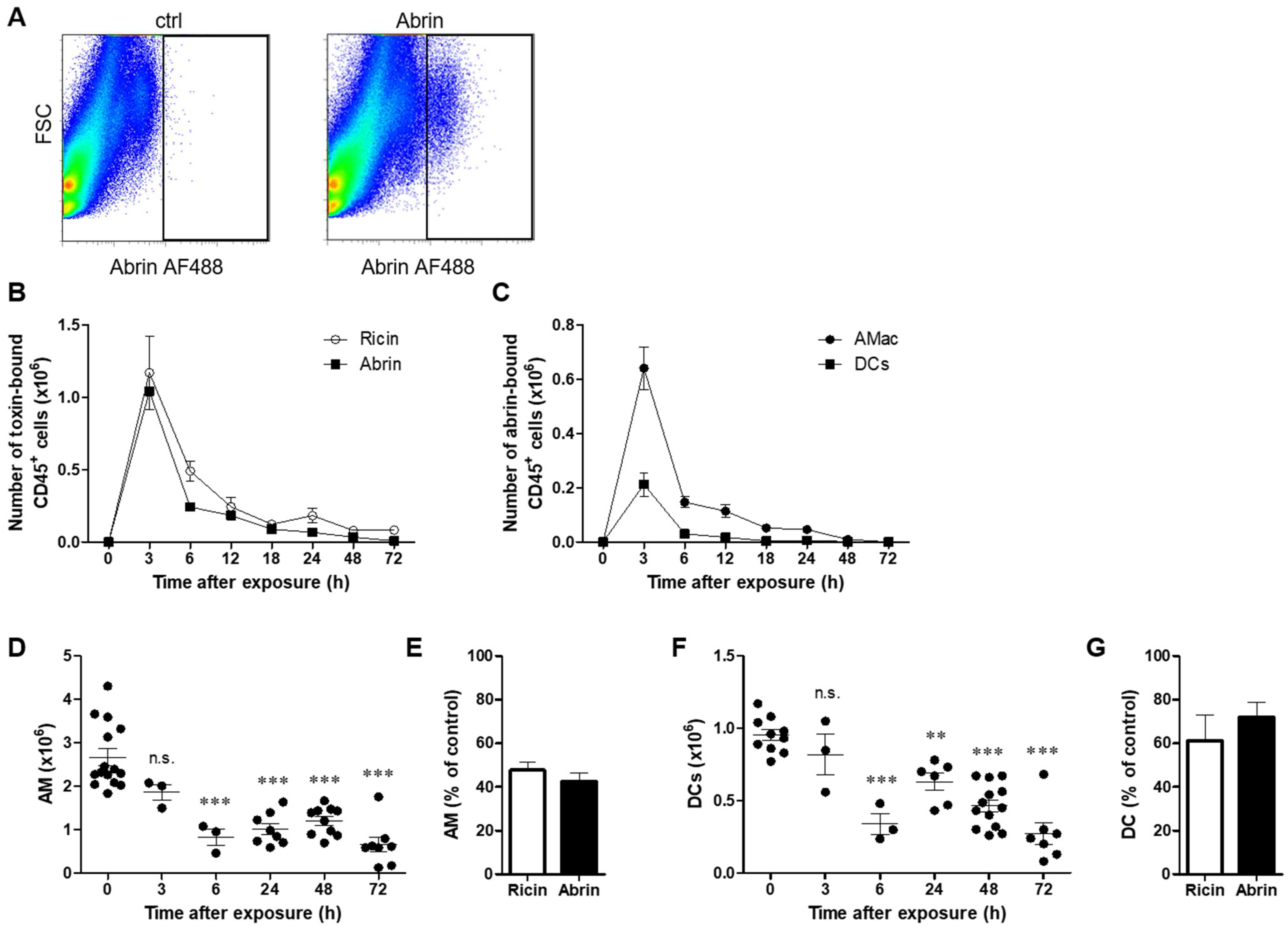
2.1. Differential Binding of Abrin to Lung Cell Populations
2.2. Pulmonary Exposure to Abrin and Ricin Induces Comparable Neutrophil Influx to the Lungs Accompanied by Lung Hyperpermeability
2.3. Pulmonary Exposure to Abrin Leads to Inferior Impairment of Junction Proteins in the Lungs in Comparison to Ricin
2.4. Pulmonary Exposure to Abrin- and Ricin-Induced Comparable Damage to the Endothelial Glycocalyx
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Fluorescent Toxin Labeling and Intoxication
4.3. Flow Cytometry
4.4. Immunohistochemistry
4.5. Permeability Analysis
4.6. Analysis of Glycocalyx Shedding
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Maro, A.; Citores, L.; Russo, R.; Iglesias, R.; Ferreras, J.M. Sequence comparison and phylogenetic analysis by the Maximum Likelihood method of ribosome-inactivating proteins from angiosperms. Plant Mol. Biol. 2014, 85, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Landi, N.; Ruocco, M.R.; Ragucci, S.; Aliotta, F.; Nasso, R.; Pedone, P.V.; Di Maro, A. Quinoa as source of type 1 ribosome inactivating proteins: A novel knowledge for a revision of its consumption. Food Chem. 2021, 342, 128337. [Google Scholar] [CrossRef] [PubMed]
- Stirpe, F.; Gilabert-Oriol, R. Ribosome-Inactivating Proteins: An Overview; Plant Toxins; Carlini, C.R., Ligabue-Braun, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 153–182. [Google Scholar]
- Girbés, T.; Ferreras, J.M.; Arias, F.J.; Stirpe, F. Description, distribution, activity and phylogenetic relationship of ribosome-inactivating proteins in plants, fungi and bacteria. Mini Rev. Med. Chem. 2004, 4, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Ng, T. Peptides and proteins from fungi. Peptides 2004, 25, 1055–1073. [Google Scholar] [CrossRef]
- Pizzo, E.; Di Maro, A. A new age for biomedical applications of Ribosome Inactivating Proteins (RIPs): From bioconjugate to nanoconstructs. J. Biomed. Sci. 2016, 23, 54. [Google Scholar] [CrossRef]
- Liu, R.S.; Yang, J.H.; Liu, W.Y. Isolation and enzymatic characterization of lamjapin, the first ribosome-inactivating protein from cryptogamic algal plant (Laminaria japonica A). Eur. J. Biochem. 2002, 269, 4746–4752. [Google Scholar] [CrossRef]
- O’Loughlin, E.V.; Robins-Browne, R.M. Effect of Shiga toxin and Shiga-like toxins on eukaryotic cells. Microbes Infect. 2001, 3, 493–507. [Google Scholar] [CrossRef]
- Lapadula, W.J.; Sanchez Puerta, M.V.; Juri Ayub, M. Revising the taxonomic distribution, origin and evolution of ribosome inactivating protein genes. PLoS ONE 2013, 8, e72825. [Google Scholar] [CrossRef]
- Sandvig, K.; Olsnes, S.; Pihl, A. Kinetics of binding of the toxic lectins abrin and ricin to surface receptors of human cells. J. Biol. Chem. 1976, 251, 3977–3984. [Google Scholar] [CrossRef]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, M.J. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Sandvig, K.; Van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [CrossRef]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [CrossRef]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Montanaro, L.; Sperti, S.; Testoni, G.; Mattioli, A. Effect of elongation factor 2 and of adenosine diphosphate-ribosylated elongation factor 2 on translocation. Biochem. J. 1976, 156, 15–23. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Didier, P.J.; Sivasubramani, S.K.; Doyle, L.A.; Holley, J.; Roy, C.J. Pathology of lethal and sublethal doses of aerosolized ricin in rhesus macaques. Toxicol. Pathol. 2014, 42, 573–581. [Google Scholar] [CrossRef]
- Griffiths, G.D.; Rice, P.; Allenby, A.C.; Bailey, S.C.; Upshall, D.G. Inhalation toxicology and histopathology of ricin and abrin toxins. Inhal. Toxicol. 1995, 7, 269–288. [Google Scholar] [CrossRef]
- Pincus, S.H.; Bhaskaran, M.; Brey, R.N., III; Didier, P.J.; Doyle-Meyers, L.A.; Roy, C.J. Clinical and pathological findings associated with aerosol exposure of macaques to ricin toxin. Toxins 2015, 7, 2121–2133. [Google Scholar] [CrossRef]
- Wilhelmsen, C.; Pitt, M. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet. Pathol. 1996, 33, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Gal, Y.; Mazor, O.; Alcalay, R.; Seliger, N.; Aftalion, M.; Sapoznikov, A.; Falach, R.; Kronman, C.; Sabo, T. Antibody/doxycycline combined therapy for pulmonary ricinosis: Attenuation of inflammation improves survival of ricin-intoxicated mice. Toxicol. Rep. 2014, 1, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Sabo, T.; Gal, Y.; Elhanany, E.; Sapoznikov, A.; Falach, R.; Mazor, O.; Kronman, C. Antibody treatment against pulmonary exposure to abrin confers significantly higher levels of protection than treatment against ricin intoxication. Toxicol. Lett. 2015, 237, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Gal, Y.; Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Elhanany, E.; Aftalion, M.; Ehrlich, S.; Kronman, C.; Sabo, T. Equal Neutralization Potency of Antibodies Raised against Abrin Subunits. Antibodies 2020, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Gal, Y.; Seliger, N.; Sabo, T.; Kronman, C. Diverse profiles of ricin-cell interactions in the lung following intranasal exposure to ricin. Toxins 2015, 7, 4817–4831. [Google Scholar] [CrossRef]
- Lindauer, M.L.; Wong, J.; Iwakura, Y.; Magun, B.E. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signaling. J. Immunol. 2009, 183, 1419–1426. [Google Scholar] [CrossRef]
- Sapoznikov, A.; Gal, Y.; Falach, R.; Sagi, I.; Ehrlich, S.; Lerer, E.; Makovitzki, A.; Aloshin, A.; Kronman, C.; Sabo, T. Early disruption of the alveolar-capillary barrier in a ricin-induced ARDS mouse model: Neutrophil-Dependent and-independent impairment of junction proteins. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L255–L268. [Google Scholar] [CrossRef]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B. Intrapulmonary delivery of ricin at high dosage triggers a systemic inflammatory response and glomerular damage. Am. J. Pathol. 2007, 170, 1497–1510. [Google Scholar] [CrossRef]
- Harris, E.S.; Nelson, W.J. VE-Cadherin: At the front, center, and sides of endothelial cell organization and function. Curr. Opin. Cell Biol. 2010, 22, 651–658. [Google Scholar] [CrossRef]
- Herrero, R.; Sanchez, G.; Lorente, J.A. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 2018, 6, 32. [Google Scholar] [CrossRef]
- Ohta, H.; Chiba, S.; Ebina, M.; Furuse, M.; Nukiwa, T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L193–L205. [Google Scholar] [CrossRef] [Green Version]
- Katalan, S.; Falach, R.; Rosner, A.; Goldvaser, M.; Brosh-Nissimov, T.; Dvir, A.; Mizrachi, A.; Goren, O.; Cohen, B.; Gal, Y. A novel swine model of ricin-induced acute respiratory distress syndrome. Dis. Models Mech. 2017, 10, 173–183. [Google Scholar] [CrossRef]
- Schmidt, E.P.; Lee, W.L.; Zemans, R.L.; Yamashita, C.; Downey, G.P. On, around, and through: Neutrophil-Endothelial interactions in innate immunity. Physiology 2011, 26, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Schmidt, E.P. The endothelial glycocalyx: An important regulator of the pulmonary vascular barrier. Tissue Barriers 2013, 1, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Curry, F.; Adamson, R. Endothelial glycocalyx: Permeability barrier and mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Torres Filho, I.P.; Torres, L.N.; Salgado, C.; Dubick, M.A. Plasma syndecan-1 and heparan sulfate correlate with microvascular glycocalyx degradation in hemorrhaged rats after different resuscitation fluids. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1468–H1478. [Google Scholar] [CrossRef]
- Lindauer, M.; Wong, J.; Magun, B. Ricin toxin activates the NALP3 inflammasome. Toxins 2010, 2, 1500–1514. [Google Scholar] [CrossRef]
- Gal, Y.; Sapoznikov, A.; Falach, R.; Ehrlich, S.; Aftalion, M.; Kronman, C.; Sabo, T. Total body irradiation mitigates inflammation and extends the therapeutic time window for anti-ricin antibody treatment against pulmonary ricinosis in mice. Toxins 2017, 9, 278. [Google Scholar] [CrossRef]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 1993, 1154, 237–282. [Google Scholar] [CrossRef]
- Falach, R.; Sapoznikov, A.; Gal, Y.; Israeli, O.; Leitner, M.; Seliger, N.; Ehrlich, S.; Kronman, C.; Sabo, T. Quantitative profiling of the in vivo enzymatic activity of ricin reveals disparate depurination of different pulmonary cell types. Toxicol. Lett. 2016, 258, 11–19. [Google Scholar] [CrossRef]
- Aulakh, G.K. Neutrophils in the lung: “The first responders”. Cell Tissue Res. 2018, 371, 577–588. [Google Scholar] [CrossRef]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Doerschuk, C.M. Leukocyte trafficking in alveoli and airway passages. Respir. Res. 2000, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-C.; Tsai, Y.-F.; Pan, Y.-L.; Hwang, T.-L. Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed. J. 2021, 44, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Shen, L. Tight junctions on the move: Molecular mechanisms for epithelial barrier regulation. Ann. New York Acad. Sci. USA 2012, 1258, 9–18. [Google Scholar] [CrossRef]
- Cohen, O.; Mechaly, A.; Sabo, T.; Alcalay, R.; Aloni-Grinstein, R.; Seliger, N.; Kronman, C.; Mazor, O. Characterization and epitope mapping of the polyclonal antibody repertoire elicited by ricin holotoxin-based vaccination. Clin. Vaccine Immunol. 2014, 21, 1534–1540. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapoznikov, A.; Gal, Y.; Alcalay, R.; Evgy, Y.; Sabo, T.; Kronman, C.; Falach, R. Characterization of Lung Injury following Abrin Pulmonary Intoxication in Mice: Comparison to Ricin Poisoning. Toxins 2022, 14, 614. https://doi.org/10.3390/toxins14090614
Sapoznikov A, Gal Y, Alcalay R, Evgy Y, Sabo T, Kronman C, Falach R. Characterization of Lung Injury following Abrin Pulmonary Intoxication in Mice: Comparison to Ricin Poisoning. Toxins. 2022; 14(9):614. https://doi.org/10.3390/toxins14090614
Chicago/Turabian StyleSapoznikov, Anita, Yoav Gal, Ron Alcalay, Yentl Evgy, Tamar Sabo, Chanoch Kronman, and Reut Falach. 2022. "Characterization of Lung Injury following Abrin Pulmonary Intoxication in Mice: Comparison to Ricin Poisoning" Toxins 14, no. 9: 614. https://doi.org/10.3390/toxins14090614