Abstract
Voltage-gated sodium channels (VGSCs, or Nav) are important determinants of action potential generation and propagation. Efforts are underway to develop medicines targeting different channel subtypes for the treatment of related channelopathies. However, a high degree of conservation across its nine subtypes could lead to the off-target adverse effects on skeletal and cardiac muscles due to acting on primary skeletal muscle sodium channel Nav1.4 and cardiac muscle sodium channel Nav1.5, respectively. For a long evolutionary process, some peptide toxins from venoms have been found to be highly potent yet selective on ion channel subtypes and, therefore, hold the promising potential to be developed into therapeutic agents. In this research, all-atom molecular dynamic methods were used to elucidate the selective mechanisms of an analgesic-antitumor β-scorpion toxin (AGAP) with human Nav1.4 and Nav1.5 in order to unravel the primary reason for the production of its adverse reactions on the skeletal and cardiac muscles. Our results suggest that the rational distribution of residues with ring structures near position 38 and positive residues in the C-terminal on AGAP are critical factors to ensure its analgesic efficacy. Moreover, the substitution for residues with benzene is beneficial to reduce its side effects.
Keywords:
voltage-gated sodium channel; Nav1.4; Nav1.5; analgesic-antitumor peptide; subtype selectivity; adverse drug reaction; molecular dynamics Key Contribution:
The pivotal components associated with the subtype selectivity of AGAP to human Nav1.4 and Nav1.5 were excavated to provide detailed information on the rational design of high-efficiency and safety peptide medicines targeting voltage-gated sodium channels.
1. Introduction
Voltage-gated sodium channels (VGSCs) play important roles in membrane excitability transduction [1]. Mammals express nine subtypes of VGSCs (Nav1.1–1.9) according to their different tissue distributions and functions. Modification of each subtype yields different biological responses [2,3]. Among these VGSCs, Nav1.4 is mainly responsible for generating action potentials in skeletal muscles. When this subtype is activated, the action potential rapidly conveys excitement through the skeletal muscle fibers and regulates the release of Ca2+ from myofibrils to drive the contraction and relaxation of skeletal muscle. Clinical and electromyographical features reveal that mutations in the encoding gene SCN4A of Nav1.4 can trigger hyperkalemic periodic paralysis and paramyotonia congenital, while sodium channel myotonias may produce gain-of-function changes [4], but loss-of-function mutations may induce hypokalamic periodic paralysis and myasthenic weakness [5]. Encoded by the gene SCN5A, Nav1.5 is the primary segment within the intercalated disks in atrial and ventricular myocytes [6]. Gain-of-function mutations in this gene are related to the disruption of fast inactivation, persistent sodium current generation, and ventricular action potential prolongation [7]. However, loss-of-function mutations in SCN5A disrupt the membrane trafficking of the channel protein. In addition, the majority of patients carrying these mutations are diagnosed with cardiac diseases such as LQT3, Brugada syndrome, and sick sinus syndrome [8,9].
In previous comprehensive studies, by inhibiting human Nav1.7 (hNav1.7) with potential analgesic effects, a kind of β-Scorpion Toxin (β-ScTx) was found and named analgesic-antitumor peptide (AGAP, Figure 1) [10,11]. Based on the genetic evidence that gain-of-function and loss-of-function mutations of this sodium channel coding gene may cause painful syndromes and pain insensitivity, Nav1.7 has emerged as a promising and well-validated pain target [12,13,14,15]. Therefore, it is reasonable to believe that this VGSC subtype, which is distributed primarily in the peripheral nervous system, is an ideal target for developing non-addictive therapeutics for pain. However, it is alarming that the off-target effects of the analgesic on Nav1.4 and Nav1.5 could contribute to the possible side effects on the skeletal and cardiac muscles, which may be due to the relatively high conservation of different VGSC subtypes. Unrestricted VGSC blockage could cause heart failure, paralysis, and respiratory failure because it impairs the activity of Nav1.4 and Nav1.5, which are the primary sodium channels in the skeletal and cardiac muscles, respectively. In fact, a part of analgesic targeting hNav1.7 has to be abandoned in its clinical trials for the above reasons [16,17,18].
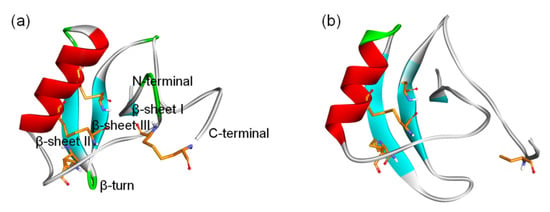
Figure 1.
3D structure of β-ScTxs. (a) The crystal structure of a toxin from the scorpion Centruroides noxius Hoffmann (2YC1); (b) The structure model of AGAP. Residues forming four disulfide bonds are orange; α-helices and β-sheets are colored red and blue, respectively. Naming notations of secondary structures are labeled in (a).
The usage of animal venom to develop novel medicines and serve as pharmacological instruments for monitoring voltage-gated sodium channel function has long been recognized. Among them, the scorpion toxins acting on VGSC can be divided into α- and β-Scorpion Toxins (ScTx) based on their differences in electrophysiological properties and binding sites. The α-ScTx binding on site 3 in VGSC is believed to trap the S4 segment on DIV in an inward position, which prevents it from moving normally in response to depolarization and prolongs action potentials. Meanwhile, β-ScTx can reduce the amplitude of the peak currents and potentiate the activation of the Na+ channels by binding to site 4 in VGSCs (Figure 2) [19,20]. For instance, Huang et al. found that an α-like scorpion toxin, OD-1, had an agonistic effect on VGSC and served as a new excitotoxicity and seizure model to explore the underlying mechanism of a novel third-generation antiepileptic drug [21,22]. Unlike that, AGAP from Buthus martensii Karsch (Bmk) is a 66-amino acid neurotoxin that belongs to β-ScTx [23]. The outcomes of our earlier electrophysiological studies on AGAP were exactly consistent with this characteristic. In the whole-cell patch clamp tests on different VGSC subtypes, compared to the control, the peak current was significantly suppressed. The analysis of the current–voltage relationship displayed a negative shift in the voltage dependence of activation following priming depolarizations after exposure to 100 nM AGAP. In contrast, no significant action on V1/2 values of inactivation was observed, and the duration of the recovery process stayed mainly unchanged. As a result, this peptide is defined as a kind of β-ScTx by this current–voltage relationship for AGAP-modified sodium currents [24]. Four homologous domains (DI–DIV) form the α-subunit of VGSC (Figure 2). Site 4 is formed by the extracellular loops connecting transmembrane helices S1–S2 and S3–S4 at DII. Likewise, the extracellular loops S1–S2 and S3–S4 are also the principal components of site 3, but this binding site is located on DIV [25]. After activation by a strong depolarization, i.e., the toxin trapped into the VGSC active conformation, the residues of S4 on the voltage sensing domain (VSD) of DII targeted by β-ScTx will be exposed [26]. Cumulatively, subsequent activation and repetitive action potential firing will be enhanced [27]. Therefore, β-ScTx can alert the action potential of VGSCs in the activated state. Extensive experimental and computational studies have provided further insights into the interaction mechanism between β-ScTx and VGSCs. These results identified that substitution of E779 and P782 at DII/S1-S2 or A841, N842, V843, E844, G845, and L846 at DII/S3-S4 reduced the bioactivity of Css4 (a kind of β-ScTx from Centruroides suffusus) on rNav1.2. Furthermore, the replacement of DII in this channel with the counterpart DmNav1 domain from Drosophila melanogaster inverted the insensitivity of AahIT (a kind of β-ScTx from Androctonus australis) to rNav1.2 [28,29].
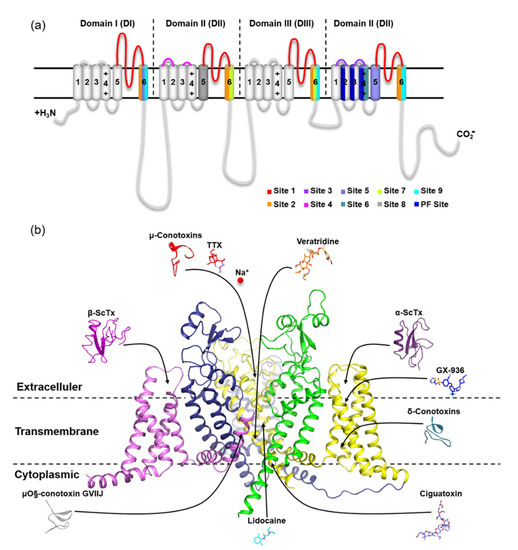
Figure 2.
Location of nine receptor sites on mammalian VGSC α subunits. (a) Topology of mammalian VGSC α subunits indicating binding sites. Representative inhibitors of sites 1–7 and 9 and the PF site on VGSC are indicated with arrows pointing to the general location of their respective primary receptor sites and colored; (b) Side view of the human Nav1.7 subunit complex structure (6J8G) in part with the VSD of DI and PM of DII removed for clarity. The representative inhibitors of site 8 (pyrethroids) are synthetic analogs of pyrethrins and are, therefore, not shown here. The PDB codes for peptide toxins are μ-conotoxin (1TCG), α-scorpion toxin (2ASC), β-scorpion toxins (2YC1), δ-conotoxin (1G1P), and μO§-conotoxin GVIIJ (2N8H).
With IC50 values of 3.25 × 10−8 M, 2.19 × 10−8 M, and 1.41 × 10−8 M, respectively, the similar biological activity of AGAP to hNav1.7, hNav1.4 and hNav1.5 is the most likely reason for the occurrence of their adverse effects on the skeletal and cardiac muscles. This possibility was confirmed in subsequent animal experiments. The measurement of heart rate, creatine kinase (CK), and lactic dehydrogenase (LDH) in mice after intravenous injection of AGAP verified the acute toxicity of this peptide to cardiac muscle and could not survive more than six days, even at a low dose of AGAP-treated group. It also led to the absence of motor function tests because no eligible mice survived in this group [30]. To address this problem, we conducted multiple cellular and molecular studies, and the results primarily identified the importance of W38 on AGAP in the specificity of different VGSC subtypes and screened two effective mutants (AGAPW38G/AGAPW38F). The whole-cell clamp patch test and in vivo experiments indicated that there were no significant effects on skeletal and cardiac muscles after intravenously injecting AGAPW38G compared with the saline-treated group [24,31]. To date, there has not been enough evidence to provide a panoramic mechanistic understanding of how AGAP interacts with different VGSC subtypes. In light of our previous results on the binding modes of AGAP and hNav1.7 [32], we herein elucidated the detailed mechanism of AGAP mutants with hNav1.4 and hNav1.5 through dynamic simulations, revealing the reason why the mutation of one single amino acid can bring about the remarkable alteration of subtype selectivity. We believe these findings are not only beneficial to avoid toxicity to the muscles and myocardium but also helpful to promote progress in developing safer and more effective treatments aimed at VGSC subtypes.
2. Results
For ease of presentation in this paper, the amino acid residues on VGSCs were indicated by three-letter abbreviations and on peptides by single-letter abbreviations. The other abbreviations were listed in the Supplementary Materials as well (Table S3).
2.1. Differences in 3D Structures of VSD2hNav1.4 and VSD2hNav1.5 in Comparison with VSD2hNav1.7
MD simulations were carried out to clarify the effects of receptor structure differences on the selectivity of AGAP. The RMSDs of VSD2s on hNav1.4, hNav1.5, and hNav1.7 during MD simulations were 0.39 nm, 0.39 nm, and 0.37 nm, respectively, indicating the similar stability of these three systems (Figure 3). As the major binding site for β-ScTx, the extracellular loops connecting DII/S1–S2 and DII/S3–S4 displayed significant primary sequence alignment identity differences with the transmembrane helices (S1–S4) (Figure 4). The different residues in S1–S3 may interact with conserved negatively charged residues in S4 to form different salt bridges (Figure 5). Specifically, R114 and R117 on S4 directly engage E40 on S1 and E98 on S3 of VSD2hNav1.7 through salt-bridge interactions so that the distances between these three helices were shortened. Similar trends were observed for R114 and E40 of VSD2hNav1.5 but not for VSD2hNav1.4. As a result, the binding site of β-ScTxs, the gap between the two extracellular loops on S1–S2 and S3–S4 of VSD2hNav1.7, is much more compact than the gap of VSD2hNav1.4 and VSD2hNav1.5. The discrepancy between the spatial structures of the active pockets may further explain the different bonding strengths of the same types of inhibitors. However, when the IC50 values were identified by patch clamp to detect the different subtype selectivity of AGAP, the results were similar [24,31]. It is, therefore, still necessary to further explore the interaction mechanism of AGAP with hNav1.4, hNav1.5, and hNav1.7.
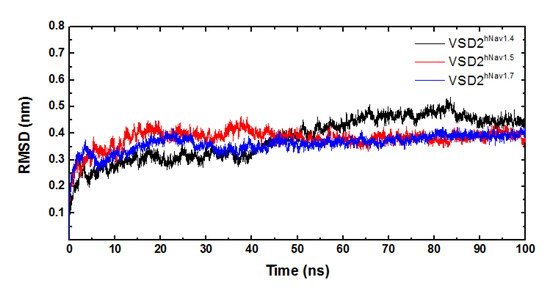
Figure 3.
RMSD curves of systems of VSD2s on hNav1.4, hNav1.5, and hNav1.7.
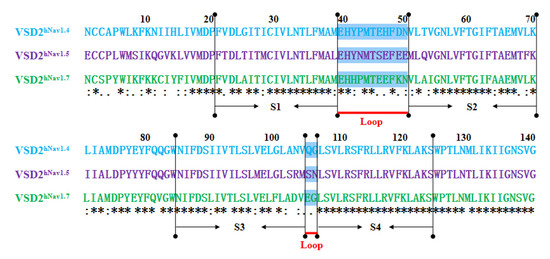
Figure 4.
Sequence alignment for VSD2s on hNav1.4, hNav1.5, and hNav1.7. “*” means the residues in this location are identical in three VGSC isoforms; “:” means similar, and “.” means a little similar. Dashed lines in green indicate the extracellular loops connecting S1–S2 and S3–S4 on VSD2s.
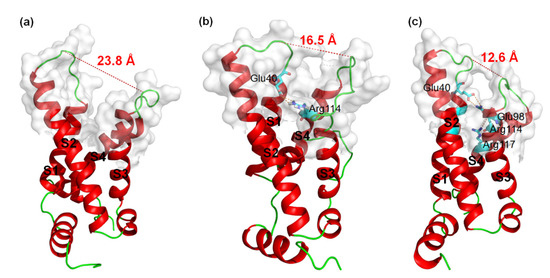
Figure 5.
3D structures of VSD2s on hNav1.4, hNav1.5, and hNav1.7. (a) VSD2hNav1.4; (b) VSD2hNav1.5; (c) VSD2hNav1.7. Dashed lines in orange indicate salt bridges; in red are widths of the active pockets formed by loops between DII S1–2 and DII S3–4; amino acid residues formed by salt bridges are in sticks.
2.2. Analysis of the Binding Modes of AGAP and the W38G/W38F Mutant with VSD2hNav1.4 and VSD2hNav1.5
From 100 ns MD simulations, the static binding poses of AGAP and its two mutants AGAPW38G/W38F with VSD2hNav1.4 and VSD2hNav1.5 were obtained. Similar to hNav1.7, the major interaction regions were located on the β-turn and C-terminal in the AGAP peptides.
2.2.1. A Structural Model for the β-ScTx-hNav1.4 Complex
Six residues in the β-turn (W38, A39, V41, Y42, G43 and N44) participate in the combination of AGAP with VSD2hNav1.4 (Figure 6a). Among these residues, W38, A39, V41, and N44 are well positioned to interact with the bond DII/S1-S2 loop, and V41, Y42, and G43 interact with the DII/S3–S4 loop. Moreover, W38 and V41 have wide ranges of interactions with VSD2hNav1.4. Specifically, W38 contacts Met37/His41/Pro43/Leu52 in the DII/S1–S2 loop, while V41 contacts Met39/Thr53 in the DII/S1–S2 loop and Arg111/Arg114 in the DII/S3-S4 loop. Additionally, K62, C63, N64, and G65 in the AGAP C-terminal are also active in binding with VSD2hNav1.4. They interact with Leu107/Ser108 in DII/S3–S4 except for K62, which interacts with Glu40 in the DII/S1–S2 loop. Apart from residues in these two regions, Y5, Y14, F15, and Y35 in wild type (WT) contributed to the bindings as well.
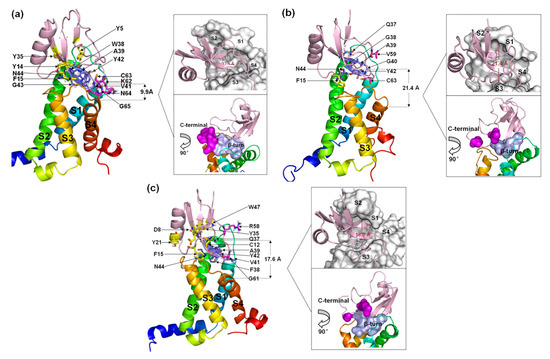
Figure 6.
Final binding poses of AGAP (a), AGAPW38G (b), and AGAPW38F (c) with VSD2 on hNav1.4. The interacting residues on the AGAP peptide are represented by sticks: purple (residues 37–44), magenta (residues 58–66), and yellow (all others). The distances between the C-terminus and DII S4 were measured. The width of the active pocket on VSD2hNav1.4 is labeled in deep red in the upper insets, which represent the top views of the complex. The interaction surfaces of the β-turn and C-terminal on AGAP/AGAPW38G/AGAPW38F with VSD2hNav1.4 are labeled purple and magenta, respectively, in the bottom insets.
When W38 is substituted in WT by G38, the number of residues with direct contact decreases dramatically (Figure 6b). In the wild-type W38 β-turn, all residues bound to VSD2hNav1.4 pointed toward the DII/S1–S2 loop. AGAPW38G failed to continue to form multiple interactions with VSD2hNav1.4. Meanwhile, V59 and C62 in the AGAPW38G C-terminus, respectively, bind to Tyr42 and Gln105 in VSD2hNav1.4. In addition, F15 also contributes significantly to binding by interacting with Arg114.
When W38 is substituted in WT by F38, six residues in the β-turn (Q37, F38, A39, V41, Y42, and N44) participate in the combination of AGAP with VSD2hNav1.4 (Figure 6c). Among these residues, Q37, F38, and A39 were positioned to interact with the DII/S1–S2 loop, while V41, Y42, and N44 interacted with the DII/S3–S4 loop. Similar to the WT, V41 in AGAP can directly contact these two extracellular loops. R58 and G61 in the AGAPW38F C-terminus combined with Tyr42 and Arg111, respectively, in VSD2hNav1.4. The interactions between D8/C12/F15 with Gln105, Y21 with Pro102, Y35/W47 with Asp49 and Met44 were also important in AGAP binding on VSD2hNav1.4.
Evidently, the binding modes of WT and AGAPW38F to hNav1.4 bear more striking resemblances compared to AGAPW38G, suggesting that the ring structure at position 38 has a critical effect on its combination with the DII/S3–S4 loop. In contrast, the interactions of the peptides with the DII/S1–S2 loop were more stable. Although the active pocket will naturally converge if the peptide contacts both extracellular loops, the inherent spacious active pocket characteristic of VSD2hNav1.4 is destined to affect the affinity of AGAP.
2.2.2. A Structural Model for the β-ScTx-hNav1.5 Complex
Seven residues in the β-turn of WT (Q37, W38, A39, G40, V41, Y42, and N44) interact directly with VSD2hNav1.5 (Figure 7a). Most of the residues were positioned to combine with the bound DII/S3–S4 loop except for Q37 and G40. Multiple interactions were observed between W38 and Glu98/Arg111/Arg114 and between N44 and Glu98/Ser102/Met104. Multiple residues in the C-terminal segments participated in the combination of the WT to DII/S3–S4 loop in VSD2hNav1.5. Moreover, Y5 and W47 in WT also contributed to the interactions with Asn43 in the DII/S1–S2 loop. On the whole, the ligand was biased to the side of the DII/S3–S4 loop.
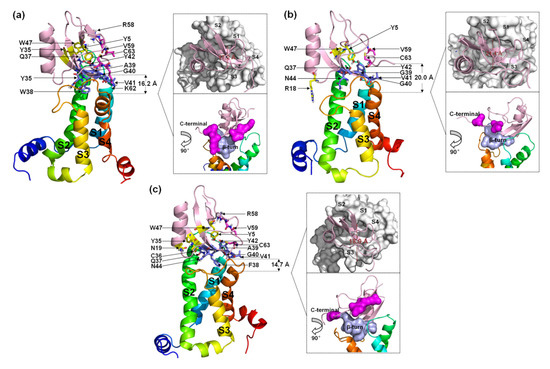
Figure 7.
Final binding poses of AGAP (a), AGAPW38G (b), and AGAPW38F (c) with VSD2 on hNav1.5. The interacting residues on the AGAP peptide are represented by sticks: purple (residues 37–44), magenta (residues 58–66), and yellow (all others). The distances between the C-terminus and DII S4 were measured. The width of the active pocket on VSD2hNav1.4 is labeled deep red in the upper insets, which represent the top views of the complex. The interaction surfaces of the β-turn and C-terminal on AGAP/AGAPW38G/AGAPW38F with VSD2hNav1.4 are labeled purple and magenta, respectively, in the bottom insets.
When W38 was substituted in WT by G38, the binding of this residue to VSD2hNav1.5 was abolished. Unlike VSD2hNav1.4, the number of residues in the β-turn that interacted with the receptor was equal to the number of residues in the β-turn that interacted with the receptor in the WT (Figure 7b). In contrast, almost all interactions between the AGAP C-terminus and VSD2hNav1.5 disappeared except for the contact between C63 and Asn106. Overall, AGAPW38G binds to the middle of the active pocket, with approximately equal distance to both of the extracellular loops.
When W38 was substituted in WT by F38, the role of this residue in combination with VSD2hNav1.5 was retained (Figure 7c). The ligand bonded with the receptor was biased to the side of the DII/S1–S2 loop due to the weakening interaction between AGAPW38F and the DII/S3-S4 loop. The function of the C-terminus is similar to the function of the WT. However, Y5, N19, Y35, and W47 are also important in peptide binding to hNav1.5. Among these residues, Y5 on β-sheet I and W47 on β-sheet II in the VSD are adjacent in space and close to Q37. These three residues may form a signature binding region to hNav1.5 compared to the other two VGSC subtypes.
The comparison of the binding modes of AGAP and the W38G/W38F mutant with hNav1.4, hNav1.5, and hNav1.7 shows that Q37, 38th, G40, V41, Y42, and N44 in the β-turn comprise a crucial binding region of the peptide when it interacts with VGSCs. Among these binding modes, Q37 is preferentially bound by DII/S1-S2 in VSD2hNav1.5, while N44 always contributes significantly to the binding of the three VGSC isoforms. The ring structure at position 38 critically affects its combination with the VGSC. Alternatively, the functions of the C-terminus in combinations with hNav1.4 and hNav1.5 are roughly identical and far less powerful than those of the C-terminus in combinations with hNav1.7. Another interesting difference is that C63 in this segment is always bound with Asn106 in VSD2hNav1.5 or the residue at position 105 in VSD2hNav1.4 and VSD2hNav1.7, which seems to indicate that the residues at this position in the different receptors have important implications for the subtype selectivity of the toxins. In addition, F15 and Y5/W47 are also important for the binding of VSD2hNav1.4 and VSD2hNav1.5, respectively.
2.3. Analysis of Dissociation Pathways of the AGAP/AGAPW38G/W38F Mutant with VSD2hNav1.4 and VSD2hNav1.5 by SMD Simulations and PMF Calculations
2.3.1. Differences in Conformations of AGAP and the W38G/W38F Mutant with VSD2hNav1.4 and VSD2hNav1.5
Based on the SMD method, the specific modes and interactions of critical residues are depicted precisely by analyzing the dissociation of the peptides from the three VGSC isoforms. The results show that AGAP, AGAPW38G, and AGAPW38F are separated completely from VSD2hNav1.4 after 3680 ps, 2950 ps, and 3360 ps (Figure 8a), as is VSD2hNav1.5 after 2880 ps, 2920 ps, and 3070 ps (Figure 8b). PMF indicates that the binding free energy of AGAP, AGAPW38G, and AGAPW38F are 190.64 kJ·mol−1, 175.68 kJ·mol−1, and 150.53 kJ·mol−1 to hNav1.4 (Figure 9a), as well as 164.81 kJ·mol−1, 146.19 kJ·mol−1, and 149.48 kJ·mol−1 to hNav1.5 (Figure 9b). Apparently, WT has a higher affinity for hNav1.4 and hNav1.5 than the two mutants. Moreover, the same change trend is expressed between dissociation time and binding affinity. Overall, the simulation and previous patch clamp experimental results are consistent with each other. Furthermore, the proportion of electrostatic and VDW interactions in AGAP binding with hNav1.5 were found not as regular as it was with hNav1.4 and hNav1.7, which were dominated by only one single type of interaction (see Section 2.3.2 and Section 2.3.3 for details).
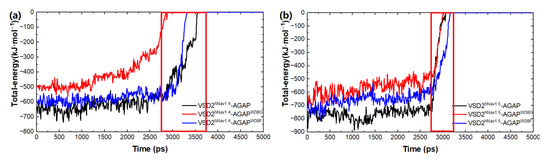
Figure 8.
Total energy of AGAP and its mutants with VSD2s on hNav1.4 (a) and hNav1.5 (b).
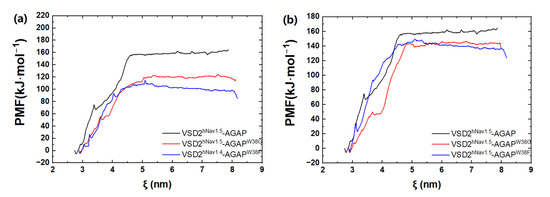
Figure 9.
PMF curves of AGAP and its mutants with VSD2s on hNav1.4 (a) and hNav1.5 (b). ζ represents the reaction coordinate generated by the configurations.
According to the comparison between the conformations of the peptides about dissociation from the receptors (Figure 10) with binding modes (Figure 6 and Figure 7), the interactions of W38 in the β-turn with DII/S1-S2 and K62 in the C-terminal with negatively charged residues in DII/S4 contribute significantly to the combination of WT to hNav1.4 and hNav1.7, but not to hNav1.5. However, mutations of position 38 can partially decrease the direct connection between G38/F38 and DII/S1–S2 but completely destroy it between the C-terminal and II/S4. Moreover, a broad binding region of β-turns to the three VGSC subtypes ensures stable and strong contact between the toxins and receptors. The interactions of the more flexible C-terminal to the VGSCs are easily reformed.
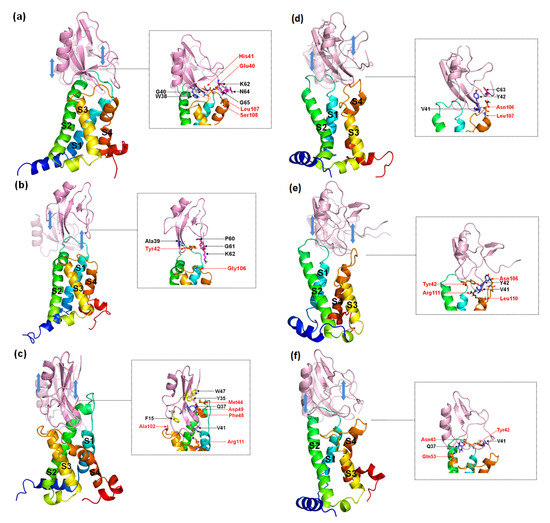
Figure 10.
Representative conformations of AGAP (a,d), AGAPW38G (b,e), and AGAPW38F (c,f) with VSD2s on hNav1.4 (a–c) and hNav1.5 (d–f) during the dissociation process. These conformations reflected the state of the receptor and the ligand just prior to complete separation. The ligands in translucency are conformations before pulling. The insets depict the interaction between the receptor and the ligand when they were to be separated. Ligand residues on the β-turn are purple, and those at the C-terminal are magenta; key residues at the interface are marked black. VSD2 hNav1.5 residues are orange and are highlighted in red characters.
2.3.2. Specific Types of Interactions of Important Residues in the β-ScTx-hNav1.4 Complex
The decompositions of the binding free energy of ligand–residue pair interactions are employed to investigate how critical components influence the affinity and selectivity of the peptides of different VGSC subtypes. The calculation results show that the average energy contributions in van der Waals (VDW) during the dissociations of AGAP, AGAPW38G, and AGAPW38F from VSD2hNav1.4 are −215.77 kJ·mol−1, −135.54 kJ·mol−1, and -187.15 kJ·mol−1, respectively, whereas in electrostatic interactions, they are −185.63 kJ·mol−1, −100.74 kJ·mol−1, and −178.81 kJ·mol−1, respectively. It follows that VDWs bear greater responsibility than electrostatic interactions to the combinations. In contrast, our previous study indicated that the latter interaction type is the dominant factor leading to the differences in the binding free energy of the three peptides to VSD2hNav1.7.
In particular, four residues (W/G/F38, A39, N42, and N44) in the β-turn play vital roles in VSD2hNav1.4 trapping (Table 1, Figure S1 and Table S1). In accordance with hNav1.7, substitution W38G strongly diminished the toxin binding affinity due to steric hindrance and H-bond repulsion between this residue and DII/S1–S2. Y42 in WT accepts a π-cation contact from Arg111 in DII/S4, which in mutants is H-bonded with DII/S3–S4 for identical contribution. N44 in WT forms weak H-bonds with the two extracellular loops, which are strong with one loop in mutants. A residue located on the loop between α-helix and β-sheet I, F15, also significantly contributes by forming a π-cation contact with the highly conserved Arg114 in DII/S4. Interestingly, the important function of F15 is to work only when the toxins are bound with hNav1.4. Therefore, we deduced that this particularity is attributed to the discrepancy in the 3D structures of the three VGSC subtypes. Additionally, unlike hNav1.7, residues in the C-terminus lack powerful interactions with hNav1.4, although they are involved in the combination of the receptor and toxins.

Table 1.
Average total-residue interaction of AGAP and its mutants with VSD2s on hNav1.4 during the dissociation process.
2.3.3. Specific Types of Interactions of Important Residues in the β-ScTx-hNav1.5 Complex
The calculation results show that the average energy contributions in van der Waals (VDW) during the dissociations of AGAP, AGAPW38G, and AGAPW38F from VSD2hNav1.5 are −169.03 kJ·mol−1, −167.40 kJ·mol−1, and −218.49 kJ·mol−1, respectively, whereas in electrostatic interactions, they are −267.36 kJ·mol−1, −158.96 kJ·mol−1, and −174.17 kJ·mol−1, respectively.
Specifically, seven residues (Q37, A39, G40, V41, N42, G43, and N44) in the β-turn have significant contributions to electrostatic interactions in all complexes (Table 2, Figure S2 and Table S2). Of these, N44 can always accept connection with DII/S3-S4. Similar to hNav1.4 and hNav1.7, the ring structure at position 38 still has a critical effect on the combination with hNav1.5. Notably, Y5 and W47, which are adjacent to each other and Q37 in space, always keep in direct contact with Asn43 in DII/S1–S2. Moreover, the residues in the DII/S1-S2 contact with Q37 in the peptide are also close to Asn43. Therefore, we inferred that the interaction surface formed by Y5, Q37, and W47 reacts with the new characteristics of the binding pose of the toxins with hNav1.5. Further analysis reveals that the production of the feature residues is derived from the opposite distribution of the hydrophobicity of the extracellular loop in DII/S1-S2 on hNav1.5 and hNav1.4/hNav1.7 (Figure 11). Because it is located in the middle of this loop on VSD2hNav1.5, hydrophilic Asn43 is able to directly contact Y5 and W47 on AGAP through electrostatic interactions. Likewise, the roles of residues in the C-terminus are limited in the combination of VSD2hNav1. 5 and toxins. Although the negatively charged R58 in this region can contact the positively charged Asn43 in DII/S1–S2, the contribution is small due to the remote distance between them.
Table 2.
Average total-residue interaction of AGAP and its mutants with VSD2s on hNav1.5 during the dissociation process.
Table 2.
Average total-residue interaction of AGAP and its mutants with VSD2s on hNav1.5 during the dissociation process.
| Region | Residue | Total Interaction (kJ·mol−1) | ||
|---|---|---|---|---|
| AGAP | AGAPW38G | AGAPW38F | ||
| β-sheet I | Y5 * 1 | −17.07 | −15.63 | −28.23 |
| loop between α-helix and β-sheet I | R18 | - | −2.83 | - |
| N19 | - | - | −35.77 | |
| β-sheet III | Y35 | −11.30 | - | −48.22 |
| C36 | - | - | −8.85 | |
| β-turn | Q37 * | −37.77 | −44.46 | −38.32 |
| W38 | −83.59 | - | - | |
| G38 | - | - | - | |
| F38 | - | - | −29.71 | |
| A39 * | −10.93 | −10.09 | −11.77 | |
| G40 * | −25.21 | −31.28 | −29.44 | |
| V41 * | −16.80 | −36.36 | −25.75 | |
| Y42 * | −23.21 | −38.87 | −16.02 | |
| N44 * | −64.25 | −24.25 | −45.47 | |
| β-sheet II | W47 * | −31.40 | −23.19 | −19.55 |
| C-terminal | R58 | −19.23 | - | −8.82 |
| V59 | −5.30 | −2.39 | −2.30 | |
| K62 | −33.93 | - | - | |
| C63 | −21.27 | −14.85 | −1.11 | |
1 The characters in “*” played important roles in all three systems.

Figure 11.
Hydrophobicity of the loop between S1-S2 on VSD2s on VGSCs. (a) hNav1.4; (b) hNav1.5; (c) hNav1.7. The default color spectrum used is blue–white–brown. Surfaces in blue correspond to hydrophilic residues, whereas surfaces in brown correspond to hydrophobic residues. Residues at positions 40–46 on VSD2s are shown on the surface.
Figure 11.
Hydrophobicity of the loop between S1-S2 on VSD2s on VGSCs. (a) hNav1.4; (b) hNav1.5; (c) hNav1.7. The default color spectrum used is blue–white–brown. Surfaces in blue correspond to hydrophilic residues, whereas surfaces in brown correspond to hydrophobic residues. Residues at positions 40–46 on VSD2s are shown on the surface.

3. Discussion
Combined with our previous study [32] and this research, two major factors are highly related to the selectivity of AGAP and its mutants to hNav1.4, hNav1.5, and hNav1.7. First, there are progressive dissimilarities in the 3D structures of AGAP bound, in which VSD2hNav1.7 is the narrowest, VSD2hNav1.5 is wider, and VSD2hNav1.4 is the widest. These differences in the binding poses established the feature residues of AGAP binding. Second, the affinity of WT AGAP to VGSCs is always higher than the affinity of the two mutants, according to the binding energy calculated by simulations and the IC50 values detected by experiments. This evidence fully demonstrates that the residue at position 38 on AGAP is one of the dominant factors affecting the selectivity of AGAP to the three VGSC subtypes. Moreover, the results indicate that the ring structure of this position is presented as the hinge structure, which provides a significant contribution when it contacts the channels through VDW interactions. Similarly, K62 is also the pivotal residue in the AGAP C-terminal, which may offer an impressive energy contribution to the negatively charged residue on VGSCs when salt bridges form. Benefiting from the narrow active pocket of AGAP binding to VSD2hNav1.7, K62 can contact negatively charged Asp103 and Glu105 in the DII/S3-S4 loop through powerful electrostatic interactions.
Additionally, a significant correlation was found between W38 and K62. For intermolecular interactions, these two residues displayed a synergistic effect to determine the selectivity of AGAP to different VGSC subtypes. For instance, although the affinity of W38 and F38 is similar to the affinity of hNav1.4, the binding free energy of WT AGAP is much higher than that of AGAPW38F because of the formation of a salt bridge between K62 on WT and Glu40 on VSD2hNav1.4. In contrast to AGAPW38F, the contribution of position 38 on AGAPW38G is minimal, while the contribution of K62 is still considerable when the toxin contacts VSD2hNav1.7. Finally, the affinities of the two mutants to hNav1.7 are equivalent. For intramolecular interactions, the function of K62 is affected by the substitution of residue at position 38 as well. The electrostatic interactions between K62/C63 and the negatively charged residues in the DII/S3–S4 loop on VSD2hNav1.7 were discovered to arise from the existence of the internal reaction chain on WT (C63-C12-D8-N11-R58-Y42), which limits the swing of the C-terminus. On AGAPW38F, this chain is broken because of the abolition of the interaction between Y42 and R58 by A39. In WT and AGAPW38G, the group of A39 interacting with Y42 is occupied by the combination with the receptor. Likewise, the formation of the salt bridge between K62 and the negatively charged residue in the DII/S1-S2 loop on VSD2hNav1.4 comes from a similar internal chain on WT (C63-C12-D8-N11-G61-Y42). After the mutation, the electrostatic interaction between Y42 and G61 is distributed because the reaction group in the former is occupied by A13 and Y14. The reason is the swerve of the benzene on Y42 because of the interactions between it and Gln105/Arg111 on VSD2hNav1.4. In the meantime, the shortage of the internal reaction chain to restrict the direction of the C-terminal on AGAP results in the modest energy contribution of this region when it is bound with hNav1.5.
However, there is still a sensitive difference in the affinity of the residue at position 38 to hNav1.5 on WT, AGAPW38G, and AGAPW38F. This result highlights that a single factor is not enough to determine the selectivity of AGAP to the three VGSC subtypes.
Additionally, some limitations of this study should not be omitted. Firstly, following the change of membrane potential, there are three different statuses: open, inactive, and closed state. Herein, the structures in an open state were selected to explore the interactions between AGAP and different VGSC subtypes. However, the combination of toxins and sodium channels should be dynamically regulated. For β-ScTx, a voltage sensor trapping model is universally accepted. In this model, the toxin first attaches to its receptor site on VGSC in its inactive state, leading to a concentration-dependent decrease of the peak current. As the channel is activated by a strong depolarization, a new binding site on the channel to β-ScTx is exposed due to the change of conformation of VGSC. Finally, the tightly bound toxin traps the activated conformation of the sodium channel in a process independent of unimolecular concentration [27,33]. This could possibly be the reason for parts of the studies exploring β-ScTx with activated state VGSC through computational and/or experimental approaches [34,35]. For this work, as mentioned above, the outcoming of the patch clamp test manifested that there was little effect of AGAP on the inactivation process [24]. In this way, AGAP appears to suppress the sodium channels, mainly in the open state. Therefore, the interactions between this peptide and activated VGSC were explored in our study. Nevertheless, it does not mean that the complete dynamic process of the toxin binding to the channels is fully considered. These variables should be taken into account in our further studies. Secondly, AGAP draws our attention because of its analgesic effect targeting hNav1.7. Furthermore, its potential skeletal and cardiac muscle toxicity, as well as the similar biological activity to hNav1.7, hNav1.4, and hNav1.5, led us to pay more attention to the interactions between AGAP and these three VGSC subtypes. After intravenous injection of AGAP, central nervous system diseases such as epilepsy and migraine were not observed in mice, so the combinations of AGAP with the VGSC subtypes mainly distributed in this region (Nav1.1, Nav1.2, Nav1.3, and Nav1.6) were not examined in this study. On the other hand, Nav1.7, Nav1.8, and Nav1.9 are primarily expressed in the peripheral nervous system, and all have the potential to be a non-addictive analgesic target. Our previous studies by whole-cell clamp patch indicated that AGAP could only suppress the activity of hNav1.8 with a 25% reduction in peak current while hNav1.7 with a reduction in 68% [24]. Limited by the experimental materials, the inhibiting effect of AGAP on hNav1.9 were not discussed in our research. In future investigations, the mechanism of this toxin and other VGSC subtypes should be complemented through both computational simulation and experimental methods. Thirdly, the rational design scheme of AGAP proposed in this paper still needs to be tested with additional experiments. In the sequel of our study, the new mutants will be obtained by genetic engineering. Their actual effects on different VGSC subtypes should be illustrated through whole-cell clamp patch or animal experiments, such as forced swimming test, rota-rod test, and mouse-twisting model.
4. Conclusions
Overall, the in-depth simulation analysis revealed several significant commonalities and differences in AGAP binding on the three VGSC subtypes. In general, retaining the ring structure of the amino acid residue at or near position 38, as well as increasing the rational distribution of basic residues in the C-terminal on AGAP, are indicated to be advantageous for improving the affinity of hNav1.7, which was disclosed in our previous studies [32]. In contrast, disruption of the ring structure of F15 and Y5/W47 on AGAP effectively reduced its binding activity for hNav1.4 and hNav1.5.
To elucidate these findings more specifically, from the perspective of AGAP: (i) β-turn is the essential region of AGAP to combine with the VGSC because the majority of the binding residues (at position 37–44) are located within it; (ii) the conserved N44 in the β-turn is always H-bonded to the DII/S3-S4 on the three VGSCs with a prominent energy contribution; (iii) the deficiency of the carbonyl in the R group at position 105 on the DII/S3-S4 loop of hNav1.4 and hNav1.5 may dramatically decrease its electrostatic contacts with Y42 in the β-turn or K62 in the C-terminal on the AGAP; (iv) the unique residues on AGAP may function vitally when combined with different VGSC subtypes, for example, F15 for hNav1.4 and Y5, Q37 and W47 for hNav1.5. From the perspective of three VGSC subtypes: (i) The charged residue at position 49 on VSD2 always accepts a π-cation contact with the residues bearing ring structure (Y35, W38, and Y42) in the β-turn of AGAP; (ii) The highly conserved negatively charged residues in DII/S4 (Arg111 and/or Arg114) participate in the combination with the peptides by forming H-bond or π-cation interactions with G40 and/or V41 in the AGAP β-turns. This response is in accordance with the voltage sensor trapping model, whereby the activated conformation of VSD2, in which DII/S4 moves outward, is trapped by β-ScTxs through strong binding with it. Further research should be undertaken to verify the above results based on experimental methods such as animal toxicity test, western blotting, blood assays, and clamp patch.
We believe that the enlargement of the study of the interaction mechanism of AGAP with hNav1.4, hNav1.5, and hNav1.7 will shed more light on elaborating the selectivity of scorpion toxins to different VGSC subtypes. Furthermore, this research provides more abundant theoretical knowledge and references for developing selective medicines targeting VGSCs.
5. Materials and Methods
5.1. Homology Modeling and Molecular Docking
The theoretical model of hNav1.4 was obtained from the Protein Data Bank (PDB ID: 6AGF). Five hundred conformations of hNav1.5 and AGAP were built through Modeler 9.9 [36] due to the protein sequences obtained from UniProt (accession numbers were Q15858 and Q95P69, respectively) [37]. Crystal structures of cardiac sodium channel from Rattus norvegicus (PDD ID: 6UZ0) and α-Tx11 from Buthus martensii (PDB: 2KBH) were selected as the templates for homology modeling because of their high sequence identity with hNav1.5 and AGAP, respectively. Models with the least discrete optimized protein energy (DOPE) score were validated by Ramachandran plots and profile-3D and chosen as the best ones. Two mutants of AGAP (AGAPW38G/AGAPW38F) were obtained by modeler site-directed mutagenesis. To accurately predict the three-dimensional structures of the target sequences in a real physiological environment, molecular dynamics (MD) simulations were applied. The parameters used here are described in Section 5.2.
The optimized structures of AGAP and its mutants were docked into the binding sites on VSD2hNav1.4 and VSD2hNav1.5 by ZDOCK, which is suitable for studying the interactions between biomacromolecules [38]. The active pockets were restricted in the extracellular region of VSD2. The matching algorithms are used to carry out the process of docking. In accordance with the RMSD cutoff, 2000 poses were divided into 60 clusters with an angular step size of 6°. The small angular step size ensures that the most possible binding modes can be considered in the docking. The cluster containing the largest number of docking poses usually figures out the binding site that the ligand is most likely to combine. Furthermore, the configurations with the lowest binding energy in the largest cluster were screened out for further simulations after excluding those in direct conflict with the position of the membrane and published research results about the binding site of β-ScTx with VGSC [20,28,29]. However, the molecular docking was carried out in a vacuum, which is insufficient to reflect the realistic conformations of AGAP with VGSCs. Therefore, molecular dynamic simulations were needed to optimize these conformations.
5.2. Molecular Dynamics
The prediction structures and docking complexes were carried out on 100 ns time scale molecular dynamics simulations utilizing the GROMACS 2018 package [39]. A 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer model was used with a united-atom force field [40] to describe the phospholipid bilayer of hNav1.4 and hNav1.5, and GROMOS-53a6 force-field parameters were assigned to the other parts in the systems. InflateGro methodology [41] was performed to accurately embed these two channels into the lipids. The SPC water model [42] was introduced as the solvation, and counterions were added to neutralize the systems. The steepest descent algorithm and conjugate gradient algorithm were used to minimize the energy and remove the bad contacts first. Subsequently, the simulation conditions were heated to 310 K using a modified Berendsen thermostat [43] with non-hydrogen solute atoms restrained. Simulation in the NPT ensemble follows this desired temperature and 1 atm constant pressure for 1 ns. The equilibration systems were subjected to a 100 ns MD simulation with no constraints applied. Moreover, the particle mesh Ewald (PME) method [44] and LINear Constraint Solver (LINCS) [45] were performed to assess the long-range electrostatic interactions and redress the lengths of all the bonds. The MD trajectory and snapshots were saved every 10 ps and analyzed by the tools of the Gromacs package, PyMol [46], and VMD [47]. The equilibrated trajectory was extracted to perform cluster analysis using the Gromos clustering algorithm with a tolerance of 0.15 nm for root–mean–square deviation (RMSD) [48].
5.3. Steered Molecular Dynamics and PMF Calculations
Steered molecular dynamics (SMD) simulations were carried out on the equilibrated model after MD [49]. With a biasing force constant of 500 kJ·mol−1·nm−2 and a pulling velocity of 0.001 nm·ps−1, a force pulled AGAP/AGAPW38G/AGAPW38F away from the binding surface of VSD2hNav1.4 and VSD2hNav1.5 along the z-dimension in the 5 ns simulation runs at 310 K and 1 atm. Herein, with respect to the atoms of VSD2s, the β-ScTxs were taken to be static. Snapshots of the model were captured every 10 ns. The umbrella sampling method, weighted histogram analysis method [50], and PMF (potential of mean force) curves for this process were calculated to describe the binding free energy between the ligand and receptor.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/toxins15010033/s1, Figure S1: VDW interaction and electrostatic interaction of residues on AGAP and its mutants that directly contact with VSD2s on hNav1.4; Figure S2: VDW interaction and electrostatic interaction of residues on AGAP and its mutants that directly contact with VSD2s on hNav1.5; Table S1: Residues involved in the formations of hydrogen bonds in SMD simulation of AGAP and its mutants with VSD2s on hNav1.4; Table S2: Residues involved in the formations of hydrogen bonds in SMD simulation of AGAP and its mutants with VSD2s on hNav1.5; Table S3: Abbreviations in the paper.
Author Contributions
Conceptualization, F.Z. and Y.S.; software, F.Z. and L.F.; validation, W.X. and Y.S.; formal analysis, F.Z., L.F., Q.W. and Q.Y.; investigation, Q.Y. and Y.H.; resources, F.Z., W.X. and Y.S.; data curation, F.Z., L.F., Q.W. and Q.Y.; writing—original draft preparation, F.Z. and L.F.; writing—review and editing, F.Z., W.X. and Y.S.; visualization, L.F. and Q.W.; supervision, W.X. and Y.S.; project administration, W.X. and Y.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China [81973227], the Basic Scientific Research Project of Colleges and University of Liaoning Provincial Department of Education [LJKZ0936, LJKMZ20221359], and Liaoning Scientific Provincial Nature Fund Guidance Plan [2019-ZD-0462].
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Catterall, W.A. Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 1992, 72 (Suppl. S4), 15–48. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.A.; Strege, P.R.; Tester, D.J.; Locke, G.R., III; Talley, N.J.; Bernard, C.E.; Rae, J.L.; Makielski, J.C.; Ackerman, M.J.; Farrugia, G. Sodium channel mutation in irritable bowel syndrome: Evidence for an ion channelopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G211–G218. [Google Scholar] [CrossRef]
- Puntmann, V.O.; Taylor, P.C.; Barr, A.; Schnackenburg, B.; Jahnke, C.; Paetsch, I. Towards understanding the phenotypes of myocardial involvement in the presence of self-limiting and sustained systemic inflammation: A magnetic resonance imaging study. Rheumatology 2010, 49, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Ye, J.; Tang, S.; Wang, J.; Luo, B.; Ji, F.; Zhang, X.; Yu, Y.; Cheng, X.; Li, Y. N1366S mutation of human skeletal muscle sodium channel causes paramyotonia congenita. J. Physiol. 2017, 595, 6837–6850. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.C. Channelopathies of skeletal muscle excitability. Compr. Physiol. 2015, 5, 761–790. [Google Scholar] [PubMed]
- Martin, B.; Gabris, B.; Barakat, A.F.; Henry, B.L.; Giannini, M.; Reddy, R.P.; Wang, X.; Romero, G.; Salama, G. Relaxin reverses maladaptive remodeling of the aged heart through Wnt-signaling. Sci. Rep. 2019, 9, 18545. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.; Wang, L.; Zhong, J. Sodium channels, cardiac arrhythmia, and therapeutic strategy. Adv. Pharmacol. 2014, 70, 367–392. [Google Scholar] [PubMed]
- Liu, M.; Yang, K.C.; Dudley, S.C. Cardiac Sodium Channel Mutations: Why so Many Phenotypes? Curr. Top Membr. 2016, 78, 513–559. [Google Scholar] [CrossRef] [PubMed]
- Remme, C.A.; Bezzina, C.R. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc. Ther. 2010, 28, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Ma, R.L.; Wang, S.L.; Duan, Z.Y.; Zhang, J.H.; Wu, L.J.; Wu, C.F. Expression of an antitumor-analgesic peptide from the venom of Chinese scorpion Buthus martensii karsch in Escherichia coli. Protein Expr. Purif. 2003, 27, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.H.; Ruan, J.P.; Cai, X.T.; Lu, W.G.; Ye, J.; Yang, J.; Yang, Y.; Sun, X.Y.; Cao, J.L.; Cao, P. Antinociceptive effects of analgesic-antitumor peptide (AGAP), a neurotoxin from the scorpion Buthus martensii Karsch, on formalin-induced inflammatory pain through a mitogen-activated protein kinases-dependent mechanism in mice. PLoS ONE 2013, 8, e78239. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, G.; Mcmahon, S.B. The physiological function of different voltage-gated sodium channels in pain. Nat. Rev. Neurosci. 2021, 22, 263–274. [Google Scholar] [CrossRef]
- Chew, L.A.; Bellampalli, S.S.; Dustrude, E.T.; Khanna, R. Mining the Nav1.7 interactome: Opportunities for chronic pain therapeutics. Biochem. Pharm. 2019, 163, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Loussouarn, G.; Sternberg, D.; Nicole, S.; Marionneau, C.; Le Bouffant, F.; Toumaniantz, G.; Barc, J.; Malak, O.A.; Fressart, V.; Péréon, Y.; et al. Physiological and Pathophysiological Insights of Nav1.4 and Nav1.5 Comparison. Front. Pharm. 2016, 14, 314. [Google Scholar]
- Flinspach, M.; Xu, Q.; Piekarz, A.D.; Fellows, R.; Hagan, R.; Gibbs, A.; Liu, Y.; Neff, R.A.; Freedman, J.; Eckert, W.A.; et al. Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci. Rep. 2017, 3, 39662. [Google Scholar] [CrossRef] [PubMed]
- Hagen, N.A.; Lapointe, B.; Ong-Lam, M.; Dubuc, B.; Walde, D.; Gagnon, B.; Love, R.; Goel, R.; Hawley, P.; Ngoc, A.H.; et al. A multicentre open-label safety and efficacy study of tetrodotoxin for cancer pain. Curr. Oncol. 2011, 18, e109–e116. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.R.; Tay, B.; Deuis, J.R.; Vetter, I. Sodium Channels and Venom Peptide Pharmacology. Adv. Pharmacol. 2017, 79, 67–116. [Google Scholar] [PubMed]
- De Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef]
- Lai, M.C.; Wu, S.N.; Huang, C.W. The Specific Effects of OD-1, a Peptide Activator, on Voltage-Gated Sodium Current and Seizure Susceptibility. Int. J. Mol. Sci. 2020, 21, 8254. [Google Scholar] [CrossRef]
- Hung, T.Y.; Wu, S.N.; Huang, C.W. The Integrated Effects of Brivaracetam, a Selective Analog of Levetiracetam, on Ionic Currents and Neuronal Excitability. Biomedicines 2021, 9, 369. [Google Scholar] [CrossRef] [PubMed]
- Couraud, F.; Jover, E.; Dubois, J.M.; Rochat, H. Two types of scorpion receptor sites, one related to the activation, the other to the inactivation of the action potential sodium channel. Toxicon 1982, 20, 9–16. [Google Scholar] [CrossRef]
- Xu, Y.; Meng, X.; Hou, X.; Sun, J.; Kong, X.; Sun, Y.; Liu, Z.; Ma, Y.; Niu, Y.; Song, Y.; et al. A mutant of the Buthus martensii Karsch antitumor-analgesic peptide exhibits reduced inhibition to hNa(v)1.4 and hNa(v)1.5 channels while retaining analgesic activity. J. Biol. Chem. 2017, 292, 18270–18280. [Google Scholar] [CrossRef] [PubMed]
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Cestèle, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by beta-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Pedraza Escalona, M.; Possani, L.D. Scorpion beta-toxins and voltage-gated sodium channels: Interactions and effects. Front. Biosci. 2013, 18, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Gurevitz, M. Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxicon 2012, 60, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Yarov-Yarovoy, V.; Scheuer, T.; Karbat, I.; Cohen, L.; Gordon, D.; Gurevitz, M.; Catterall, W.A. Structure-function map of the receptor site for β-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 2011, 286, 33641–33651. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, J.; Yu, Y.; Kong, X.; Meng, X.; Liu, Y.; Cui, Y.; Su, Y.; Zhao, M.; Zhang, J. Trp: A conserved aromatic residue crucial to the interaction of a scorpion peptide with sodium channels. J. Biochem. 2020, 168, 633–641. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, J.; Liu, H.; Sun, J.; Yu, Y.; Su, Y.; Cui, Y.; Zhao, M.; Zhang, J. Scorpion Toxins Targeting Voltage-gated Sodium Channels Associated with Pain. Curr. Pharm. Biotechnol. 2018, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, J.L.; Ming, H.Y.; Zhang, Y.N.; Dun, Y.Q.; Zhang, J.H.; Song, Y.B. Insights into the binding mode and functional components of the analgesic-antitumour peptide from Buthus martensii Karsch to human voltage-gated sodium channel 1.7 based on dynamic simulation analysis. J. Biomol. Struct. Dyn. 2020, 38, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Zhorov, B.S.; Du, Y.; Song, W.; Luo, N.; Gordon, D.; Gurevitz, M.; Dong, K. Mapping the interaction surface of scorpion β-toxins with an insect sodium channel. Biochem. J. 2021, 478, 2843–2869. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Conserved functional surface of antimammalian scorpion β-toxins. J. Phys. Chem. B 2012, 116, 4796–4800. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gao, B.; Peigneur, S.; Tytgat, J. How a Scorpion Toxin Selectively Captures a Prey Sodium Channel: The Molecular and Evolutionary Basis Uncovered. Mol. Biol. Evol. 2020, 37, 3149–3164. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2016, 54, 5.6.1–5.6.37. [Google Scholar]
- Consortium, U.P. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins Struct. Funct. Bioinform. 2010, 52, 80–87. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Softwarex 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Berger, O.; Edholm, O.; Jahnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef]
- Kandt, C.; Ash, W.L.; Tieleman, D.P. Setting up and running molecular dynamics simulations of membrane proteins. Methods 2007, 41, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Amira, S.; Spangberg, D.; Hermansson, K. Derivation and evaluation of a flexible SPC model for liquid water. Chem. Phys. 2004, 303, 327–334. [Google Scholar] [CrossRef]
- Lemak, A.S.; Balabaev, N.K. On The Berendsen Thermostat. Mol. Simul. 1994, 13, 177–187. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Chem. Theory Comput. 1997, 4, 1463–1472. [Google Scholar] [CrossRef]
- Makarewicz, T.; Kazmierkiewicz, R. Molecular dynamics simulation by GROMACS using GUI plugin for PyMOL. J. Chem. Inf. Model. 2013, 53, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; Van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Do, P.C.; Lee, E.H.; Le, L. Steered Molecular Dynamics Simulation in Rational Drug Design. J. Chem. Inf. Model. 2018, 58, 1473–1482. [Google Scholar] [CrossRef]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).