Protective Effects of Alginate and Chitosan Oligosaccharides against Clostridioides difficile Bacteria and Toxin

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
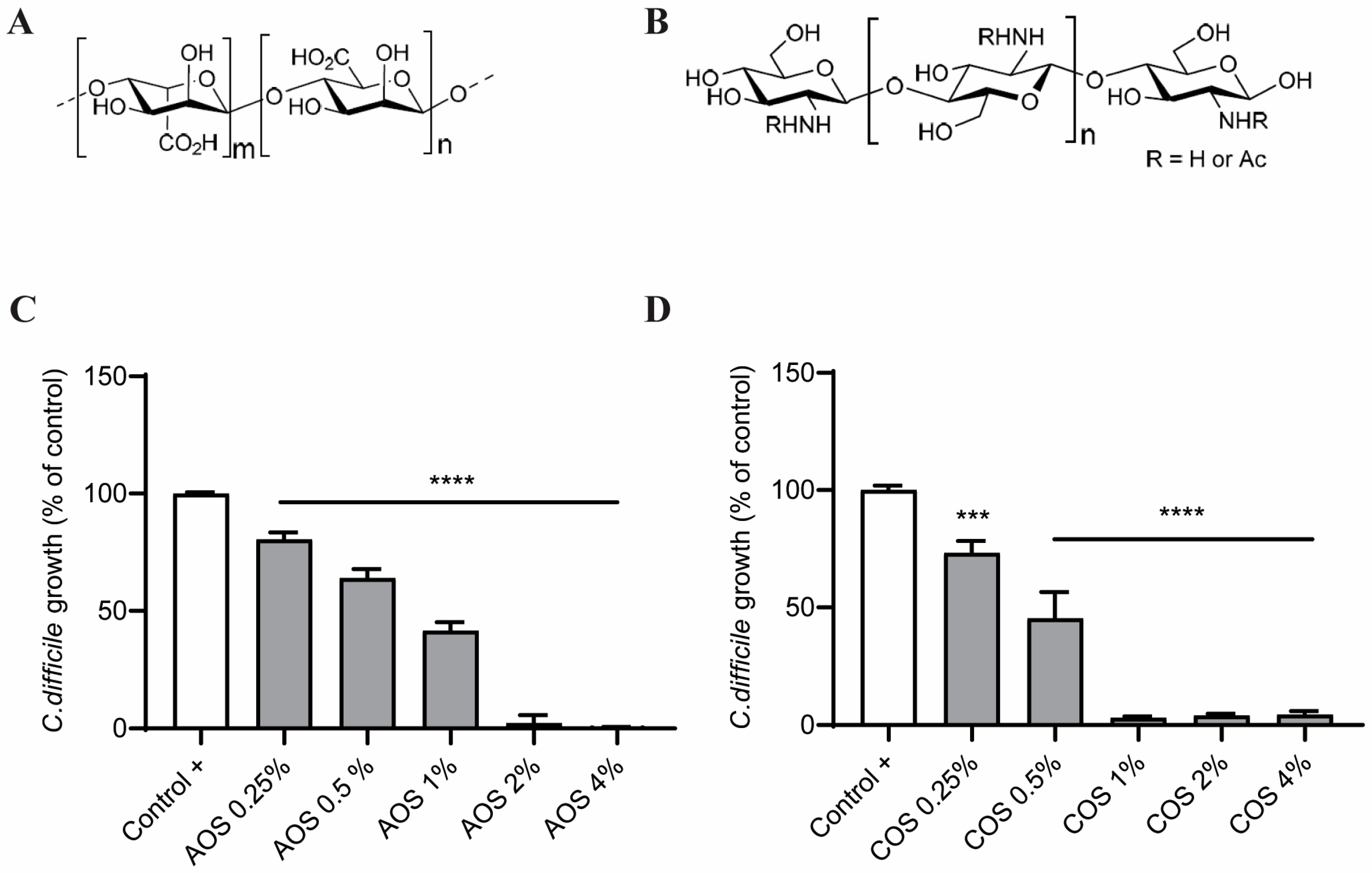
2.1. AOS and COS Reduce C. difficile Growth
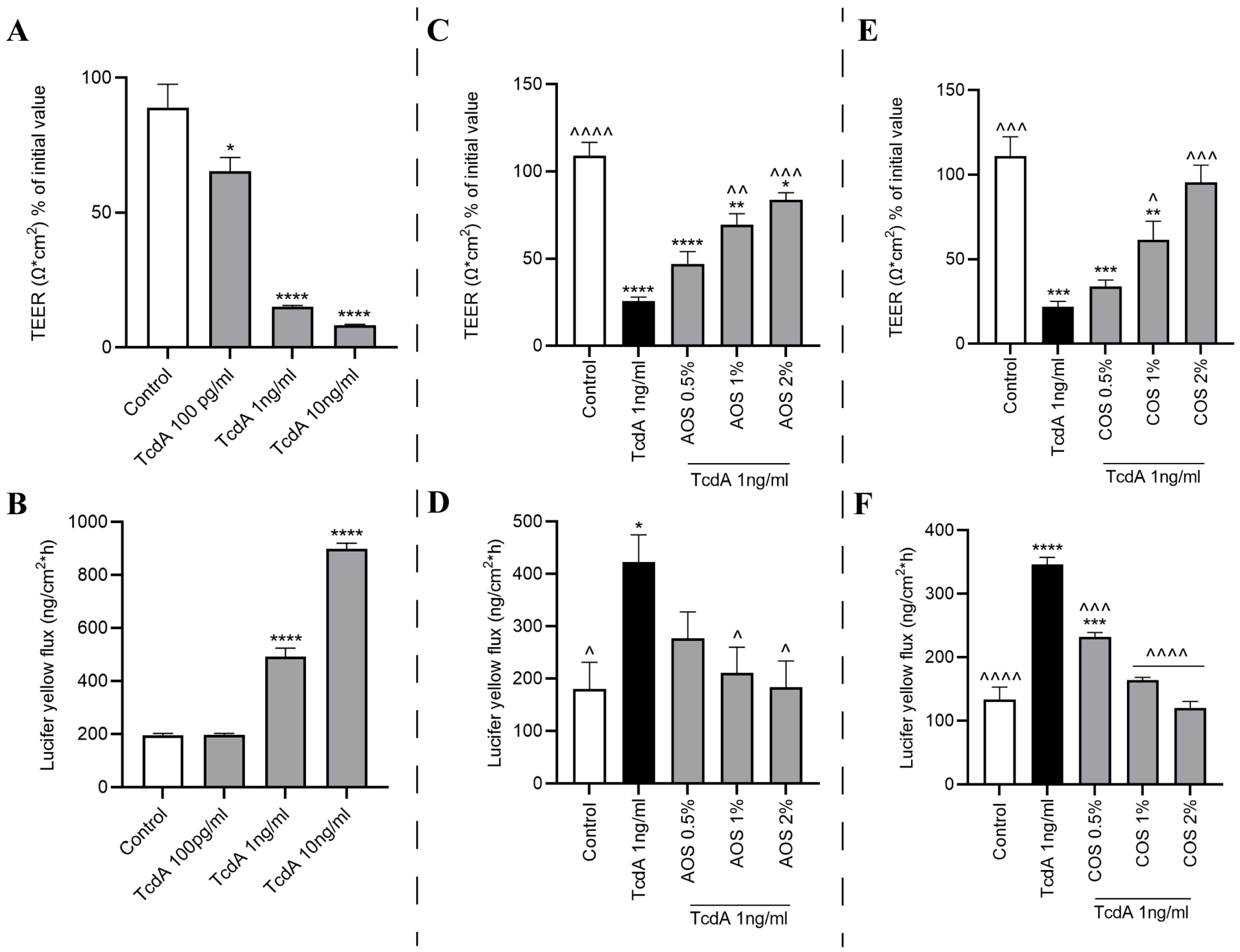
2.2. NDOs and TcdA Concentration Selection
2.3. Exposure of the Caco-2 Monolayer to TcdA Fails to Elicit IL-8 Release
2.4. Neither AOS nor COS Prevent TcdA-Induced Cell Cytotoxicity after 24 h
2.5. TcdA Impairs Caco-2 Monolayer’s Integrity
2.6. AOS and COS Possess Barrier-Protective Capability against TcdA-Induced Caco-2 Monolayer Disruption
2.7. AOS and COS Prevent the TcdA-Induced Occludin Mislocalization
2.8. Both NDOs Facilitate the Reassembly of TJs after Calcium Deprivation in Caco-2 Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Bacterial Strains and Culture Conditions
5.2. Cell Culture
5.3. Oligosaccharides and TcdA
5.4. Determination of the Minimum Inhibitory Concentration (MIC)
5.5. Cell Viability Assays
5.6. ELISA Assay for IL-8 Secretion
5.7. Transepithelial Electrical Resistance (TEER) Measurement
5.8. Paracellular Tracer Flux Assay
5.9. Calcium Switch Assay
5.10. Western Blot Analysis
5.11. Immunofluorescence Microscopy of Occludin
5.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | 5′ AMP-activated protein kinase |
| AOS | Alginate oligosaccharides |
| CaMKKβ | Calcium/Calmodulin-dependent protein kinase kinase β |
| CaSR | Calcium-sensing receptor |
| CDI | C. difficile infection |
| CDT | C. difficile transferase |
| COS | Chitin/chitosan oligosaccharides |
| DA | Degree of acetylation |
| DD | D Domain |
| DP | Degree of polymerization |
| DSS | Dextran sulfate sodium |
| E. coli | Escherichia coli |
| ELISA | Enzyme-linked immunosorbent assay |
| Gal | Galactose |
| GlcN | D-glucosamine |
| GlcNAc | N-acetyl-D-glucosamine |
| GTD | N-terminal glucosyltransferase domain |
| IP3 | D-myo-inositol 1,4,5-trisphosphate |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| LY | Lucifer yellow |
| MIC | Minimum inhibitory concentration |
| MTT | 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide |
| MW | Molecular weight |
| NDOs | Non-digestible oligosaccharides |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| RBD | Receptor binding domain |
| SCFA | Short-chain fatty acids |
| TcdA/B | TcdA/B |
| TEER | Transepithelial electrical resistance |
| TJ | Tight junction |
References
- Chandrasekaran, R.; Lacy, D.B. The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 2017, 41, 723–750. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.G.; Gerding, D.N. Clinical recognition and diagnosis of Clostridium difficile infection. Clin. Infect. Dis. 2008, 46, S12–S18. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.M.; Rathore, M.H. Clostridium difficile Infection in Children. Adv. Pediatr. 2019, 66, 263–280. [Google Scholar] [CrossRef]
- Banawas, S.S. Clostridium difficile Infections: A Global Overview of Drug Sensitivity and Resistance Mechanisms. Biomed Res. Int. 2018, 2018, 8414257. [Google Scholar] [CrossRef]
- Di Bella, S.; Ascenzi, P.; Siarakas, S.; Petrosillo, N.; Di Masi, A. Clostridium difficile Toxins A and B: Insights into Pathogenic Properties and Extraintestinal Effects. Toxins 2016, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Alyousef, A.A. Clostridium difficile: Epidemiology, pathogenicity, and an update on the limitations of and challenges in its diagnosis. J. AOAC Int. 2018, 101, 1119–1126. [Google Scholar] [CrossRef]
- Fischer, S.; Ückert, A.K.; Landenberger, M.; Papatheodorou, P.; Hoffmann-Richter, C.; Mittler, A.K.; Ziener, U.; Hägele, M.; Schwan, C.; Müller, M.; et al. Human peptide α-defensin-1 interferes with Clostridioides difficile toxins TcdA, TcdB, and CDT. FASEB J. 2020, 34, 6244–6261. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Selzer, J.; Von Eichel-Streiber, C.; Aktories, K. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J. Clin. Investig. 1995, 95, 1026–1031. [Google Scholar] [CrossRef]
- Skinner, A.M.; Phillips, S.T.; Merrigan, M.M.; O’Leary, K.J.; Sambol, S.P.; Siddiqui, F.; Peterson, L.R.; Gerding, D.N.; Johnson, S. The Relative Role of Toxins A and B in the Virulence of Clotridioides difficile. J. Clin. Med. 2020, 10, 96. [Google Scholar] [CrossRef]
- Quesada-gómez, C.; López-ureña, D.; Chumbler, N.; Kroh, H.K.; Castro-peña, C.; Rodríguez, C.; Orozco-aguilar, J.; González-camacho, S.; Rucavado, A.; Guzmán-verri, C.; et al. Analysis of TcdB Proteins within the Hypervirulent Clade 2 Reveals an Impact of RhoA Glucosylation on Clostridium difficile Proinflammatory Activities. Infect. Immun. 2016, 84, 856–865. [Google Scholar] [CrossRef]
- Pietrangeli, P.; Corpetti, C.; Seguella, L.; Del Re, A.; Pesce, M.; Vincenzi, M.; Lori, C.; Annunziata, G.; Mateescu, M.A.; Sarnelli, G.; et al. Lathyrus sativus diamine oxidase reduces Clostridium difficile toxin A-induced toxicity in Caco-2 cells by rescuing RhoA-GTPase and inhibiting pp38-MAPK/NF-κB/HIF-1α activation. Phyther. Res. 2021, 35, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, M.E.; Asadpoor, M.; Henricks, P.A.J.; Keshavarzian, A.; Folkerts, G.; Braber, S. Direct Action of Non-Digestible Oligosaccharides against a Leaky Gut. Nutrients 2022, 14, 4699. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Enteric, C.; Translational, N.; Clinic, M. The Leaky Gut: Mechanisms, Measurement and Clinical Implications in Humans. Gut 2020, 68, 1516–1526. [Google Scholar] [CrossRef]
- Christopher Peritore-Galve, F.; Shupe, J.A.; Cave, R.J.; Childress, K.O.; Kay Washington, M.; Kuehne, S.A.; Borden Lacy, D. Glucosyltransferase-dependent and independent effects of Clostridioides difficile toxins during infection. PLoS Pathog. 2022, 18, e1010323. [Google Scholar] [CrossRef]
- Du, T.; Alfa, M.J. Translocation of Clostridium difficile toxin B across polarized Caco-2 cell monolayers is enhanced by toxin A. Can. J. Infect. Dis. 2004, 15, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Folkerts, J.; Folkerts, G.; Maurer, M.; Braber, S. Microbiota-dependent and -independent effects of dietary fibre on human health. Br. J. Pharmacol. 2020, 177, 1363–1381. [Google Scholar] [CrossRef]
- Palaniappan, A.; Antony, U.; Emmambux, M.N. Current status of xylooligosaccharides: Production, characterization, health benefits and food application. Trends Food Sci. Technol. 2021, 111, 506–519. [Google Scholar] [CrossRef]
- Mrudulakumari Vasudevan, U.; Lee, O.K.; Lee, E.Y. Alginate derived functional oligosaccharides: Recent developments, barriers, and future outlooks. Carbohydr. Polym. 2021, 267, 118158. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, J.; Yin, H.; Chen, D.; Yu, B.; He, J. Ameliorative effects of alginate oligosaccharide on tumour necrosis factor-α-induced intestinal epithelial cell injury. Int. Immunopharmacol. 2020, 89, 107084. [Google Scholar] [CrossRef]
- Asadpoor, M.; Ithakisiou, G.-N.; Henricks, P.A.J.; Pieters, R.; Folkerts, G.; Braber, S. Non-digestible oligosaccharides and short chain fatty acids as therapeutic targets against enterotoxin-producing bacteria and their toxins. Toxins 2021, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Cao, Q.; Wang, Y.; Xiao, H.; Zhao, J.; Zhang, Q.; Ji, A.; Song, S. Advances in Research on the Bioactivity of Alginate Oligosaccharides. Mar. Drugs 2020, 18, 144. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Zhang, J.; Xu, Q.; Yin, H.; Chen, D.; Yu, B.; He, J. Alginate oligosaccharide protects against enterotoxigenic Escherichia coli-induced porcine intestinal barrier injury. Carbohydr. Polym. 2021, 270, 118316. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, J.; Chen, D.; Yu, B.; Mao, X.; Zheng, P.; Yu, J.; Huang, Z.; Luo, J.; Luo, Y.; et al. Alginate oligosaccharide alleviates enterotoxigenic: Escherichia coli-induced intestinal mucosal disruption in weaned pigs. Food Funct. 2018, 9, 6401–6413. [Google Scholar] [CrossRef] [PubMed]
- Muanprasat, C.; Chatsudthipong, V. Chitosan oligosaccharide: Biological activities and potential therapeutic applications. Pharmacol. Ther. 2017, 170, 80–97. [Google Scholar] [CrossRef]
- Naveed, M.; Phil, L.; Sohail, M.; Hasnat, M.; Baig, M.M.F.A.; Ihsan, A.U.; Shumzaid, M.; Kakar, M.U.; Mehmood Khan, T.; Akabar, M.D.; et al. Chitosan oligosaccharide (COS): An overview. Int. J. Biol. Macromol. 2019, 129, 827–843. [Google Scholar] [CrossRef]
- Yousef, M.; Pichyangkura, R.; Soodvilai, S.; Chatsudthipong, V.; Muanprasat, C. Chitosan oligosaccharide as potential therapy of inflammatory bowel disease: Therapeutic efficacy and possible mechanisms of action. Pharmacol. Res. 2012, 66, 66–79. [Google Scholar] [CrossRef]
- Lopez-Santamarina, A.; Mondragon, A.d.C.; Lamas, A.; Miranda, J.M.; Franco, C.M.; Cepeda, A. Animal-origin prebiotics based on chitin: An alternative for the future? A critical review. Foods 2020, 9, 782. [Google Scholar] [CrossRef]
- Guan, G.; Abul Kalam Azad, M.; Lin, Y.; Kim, S.W.; Tian, Y.; Liu, G.; Wang, H. Biological effects and applications of chitosan and chito-oligosaccharides. Front. Physiol. 2019, 10, 516. [Google Scholar] [CrossRef]
- Asadpoor, M.; Peeters, C.; Henricks, P.A.J.; Varasteh, S.; Pieters, R.J.; Folkerts, G.; Braber, S. Anti-Pathogenic Functions of Non-Digestible Oligosaccharides In Vitro. Nutrients 2020, 12, 1789. [Google Scholar] [CrossRef]
- He, N.; Yang, Y.; Wang, H.; Liu, N.; Yang, Z.; Li, S. Unsaturated alginate oligosaccharides (UAOS) protects against dextran sulfate sodium-induced colitis associated with regulation of gut microbiota. J. Funct. Foods 2021, 83, 104536. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, R.; Liu, D.; Zhang, C.; Wang, Z.A.; Du, Y. Exploring effects of chitosan oligosaccharides on the DSS-induced intestinal barrier impairment in vitro and in vivo. Molecules 2021, 26, 2199. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Zhang, J.; Chen, D.; Yu, B.; Huang, Z.; Mao, X.; Zheng, P.; Yu, J.; He, J. Alterations in intestinal microbiota by alginate oligosaccharide improve intestinal barrier integrity in weaned pigs. J. Funct. Foods 2020, 71, 104040. [Google Scholar] [CrossRef]
- Liu, P.; Piao, X.S.; Kim, S.W.; Wang, L.; Shen, Y.B.; Lee, H.S.; Li, S.Y. Effects of chito-oligosaccharide supplementation on the growth performance, nutrient digestibility, intestinal morphology, and fecal shedding of Escherichia coli and Lactobacillus in weaning pigs. J. Anim. Sci. 2008, 86, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Cheng, G.; Li, Q.; Jiao, S.; Feng, C.; Zhao, X.; Yin, H.; Du, Y.; Liu, H. Chitin oligosaccharide modulates gut microbiota and attenuates high-fat-diet-induced metabolic syndrome in mice. Mar. Drugs 2018, 16, 66. [Google Scholar] [CrossRef]
- Yang, C.M.; Ferket, P.R.; Hong, Q.H.; Zhou, J.; Cao, G.T.; Zhou, L.; Chen, A.G. Effect of chito-oligosaccharide on growth performance, intestinal barrier function, intestinal morphology and cecal microflora in weaned pigs. J. Anim. Sci. 2012, 90, 2671–2676. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, J.; Chen, D.; Yu, B.; Huang, Z.; Mao, X.; Zheng, P.; Yu, J.; He, J. Alginate oligosaccharide enhances intestinal integrity of weaned pigs through altering intestinal inflammatory responses and antioxidant status. RSC Adv. 2018, 8, 13482–13492. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, Y.; Liu, M.; Chen, L.; Meng, Q.; Tang, X.; Wang, S.; Liu, L.; Li, L.; Shen, W.; et al. Single-cell RNA sequencing analysis reveals alginate oligosaccharides preventing chemotherapy-induced mucositis. Mucosal Immunol. 2020, 13, 437–448. [Google Scholar] [CrossRef]
- Muanprasat, C.; Wongkrasant, P.; Satitsri, S.; Moonwiriyakit, A.; Pongkorpsakol, P.; Mattaveewong, T.; Pichyangkura, R.; Chatsudthipong, V. Activation of AMPK by chitosan oligosaccharide in intestinal epithelial cells: Mechanism of action and potential applications in intestinal disorders. Biochem. Pharmacol. 2015, 96, 225–236. [Google Scholar] [CrossRef]
- Shi, L.; Fang, B.; Yong, Y.; Li, X.; Gong, D.; Li, J.; Yu, T.; Gooneratne, R.; Gao, Z.; Li, S.; et al. Chitosan oligosaccharide-mediated attenuation of LPS-induced inflammation in IPEC-J2 cells is related to the TLR4/NF-κB signaling pathway. Carbohydr. Polym. 2019, 219, 269–279. [Google Scholar] [CrossRef]
- Asadpoor, M.; Varasteh, S.; Pieters, R.J.; Folkerts, G.; Braber, S. Differential effects of oligosaccharides on the effectiveness of ampicillin against Escherichia coli in vitro. PharmaNutrition 2021, 16, 100264. [Google Scholar] [CrossRef]
- Asadpoor, M.; Ithakisiou, G.N.; van Putten, J.P.M.; Pieters, R.J.; Folkerts, G.; Braber, S. Antimicrobial Activities of Alginate and Chitosan Oligosaccharides Against Staphylococcus aureus and Group B Streptococcus. Front. Microbiol. 2021, 12, 700605. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Tøndervik, A.; Sletta, H.; Klinkenberg, G.; Emanuel, C.; Onsøyen, E.; Myrvold, R.; Howe, R.A.; Walsh, T.R.; Hill, K.E.; et al. Overcoming drug resistance with alginate oligosaccharides able to potentiate the action of selected antibiotics. Antimicrob. Agents Chemother. 2012, 56, 5134–5141. [Google Scholar] [CrossRef]
- Fernandes, J.C.; Tavaria, F.K.; Soares, J.C.; Ramos, Ó.S.; João Monteiro, M.; Pintado, M.E.; Xavier Malcata, F. Antimicrobial effects of chitosans and chitooligosaccharides, upon Staphylococcus aureus and Escherichia coli, in food model systems. Food Microbiol. 2008, 25, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, J.; Gibson, G.; Formentin, K.; Beer, M.; Rastall, R. Inhibition of the adhesion of enteropathogenic Escherichia coli strains to HT-29 cells in culture by chito-oligosaccharides. Carbohydr. Polym. 2006, 64, 57–59. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Park, P.J.; Kim, S.K. Antimicrobial effect of chitooligosaccharides produced by bioreactor. Carbohydr. Polym. 2001, 44, 71–76. [Google Scholar] [CrossRef]
- Sánchez, Á.; Mengíbar, M.; Rivera-Rodríguez, G.; Moerchbacher, B.; Acosta, N.; Heras, A. The effect of preparation processes on the physicochemical characteristics and antibacterial activity of chitooligosaccharides. Carbohydr. Polym. 2017, 157, 251–257. [Google Scholar] [CrossRef]
- Laokuldilok, T.; Potivas, T.; Kanha, N.; Surawang, S.; Seesuriyachan, P.; Wangtueai, S.; Phimolsiripol, Y.; Regenstein, J.M. Physicochemical, antioxidant, and antimicrobial properties of chitooligosaccharides produced using three different enzyme treatments. Food Biosci. 2017, 18, 28–33. [Google Scholar] [CrossRef]
- Vishu Kumar, A.B.; Varadaraj, M.C.; Gowda, L.R.; Tharanathan, R.N. Characterization of chito-oligosaccharides prepared by chitosanolysis with the aid of papain and Pronase, and their bactericidal action against Bacillus cereus and Escherichia coli. Biochem. J. 2005, 391, 167–175. [Google Scholar] [CrossRef]
- Gigli, S.; Seguella, L.; Pesce, M.; Bruzzese, E.; D’Alessandro, A.; Cuomo, R.; Steardo, L.; Sarnelli, G.; Esposito, G. Cannabidiol restores intestinal barrier dysfunction and inhibits the apoptotic process induced by Clostridium difficile toxin A in Caco-2 cells. United Eur. Gastroenterol. J. 2017, 5, 1108–1115. [Google Scholar] [CrossRef]
- No, H.K.; Young Park, N.; Ho Lee, S.; Meyers, S.P. Antibacterial activity of chitosans and chitosan oligomers with different molecular weights. Int. J. Food Microbiol. 2002, 74, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Benhabiles, M.S.; Salah, R.; Lounici, H.; Drouiche, N.; Goosen, M.F.A.; Mameri, N. Antibacterial activity of chitin, chitosan and its oligomers prepared from shrimp shell waste. Food Hydrocoll. 2012, 29, 48–56. [Google Scholar] [CrossRef]
- Zheng, L.Y.; Zhu, J.F. Study on antimicrobial activity of chitosan with different molecular weights. Carbohydr. Polym. 2003, 54, 527–530. [Google Scholar] [CrossRef]
- Piotrowski, M.; Wultańska, D.; Obuch-Woszczatyński, P.; Pituch, H. Fructooligosaccharides and mannose affect Clostridium difficile adhesion and biofilm formation in a concentration-dependent manner. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.; Alston, L.; Tulk, S.E.; Schenck, L.P.; Grassie, M.E.; Alhassan, B.F.; Veermalla, A.T.; Al-Bashir, S.; Gendron, F.P.; Altier, C.; et al. The P2Y6 receptor mediates Clostridium difficile toxin-induced CXCL8/IL-8 production and intestinal epithelial barrier dysfunction. PLoS ONE 2013, 8, e81491. [Google Scholar] [CrossRef] [PubMed]
- Mahida, Y.R.; Makh, S.; Hyde, S.; Gray, T.; Borriello, S.P. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: Induction of interleukin 8 production and apoptosis after cell detachment. Gut 1996, 38, 337–347. [Google Scholar] [CrossRef]
- He, D.; Sougioultzis, S.; Hagen, S.; Liu, J.; Keates, S.; Keates, A.C.; Pothoulakis, C.; LaMont, J.T. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology 2002, 122, 1048–1057. [Google Scholar] [CrossRef]
- Shen, A. Clostridium difficile toxins: Mediators of inflammation. J. Innate Immun. 2012, 4, 149–158. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Firoozabadi, T.P.; Shankayi, Z.; Izadi, A.; Firoozabadi, S.M.P. Can Lucifer Yellow indicate correct permeability of biological cell membrane under an electric and magnetic field? Cell J. 2015, 16, 560–563. [Google Scholar] [CrossRef]
- Xiong, B.; Liu, M.; Zhang, C.; Hao, Y.; Zhang, P.; Chen, L.; Tang, X.; Zhang, H.; Zhao, Y. Alginate oligosaccharides enhance small intestine cell integrity and migration ability. Life Sci. 2020, 258, 118085. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Tian, G.; Chen, D.W.; Yao, Y.; He, J.; Zheng, P.; Mao, X.B.; Yu, J.; Huang, Z.Q.; Yu, B. Involvement of PKA signalling in anti-inflammatory effects of chitosan oligosaccharides in IPEC-J2 porcine epithelial cells. J. Anim. Physiol. Anim. Nutr. 2018, 102, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Nobile, N.; Gigli, S.; Seguella, L.; Pesce, M.; d’Alessandro, A.; Bruzzese, E.; Capoccia, E.; Steardo, L.; Cuomo, R.; et al. Rifaximin improves Clostridium difficile toxin A-induced toxicity in Caco-2 cells by the PXR-dependent TLR4/MyD88/NF-κB pathway. Front. Pharmacol. 2016, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Berkes, J.; Viswanathan, V.K.; Savkovic, S.D.; Hecht, G. Intestinal epithelial responses to enteric pathogens: Effects on the tight junction barrier, ion transport, and inflammation. Gut 2003, 52, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Hecht, G.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J. Clin. Investig. 1988, 82, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Pothoulakis, C.; Thomas Lamont, J. Protein kinase C signaling regulates ZO-1 translocation and increased paracellular flux of T84 colonocytes exposed to Clostridium difficile toxin A. J. Biol. Chem. 2002, 277, 4247–4254. [Google Scholar] [CrossRef]
- González-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 729–756. [Google Scholar] [CrossRef]
- Nusrat, A.; Von Eichel-Streiber, C.; Turner, J.R.; Verkade, P.; Madara, J.L.; Parkos, C.A. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect. Immun. 2001, 69, 1329–1336. [Google Scholar] [CrossRef]
- Allam-Ndoul, B.; Castonguay-Paradis, S.; Veilleux, A. Gut Microbiota and Intestinal Trans-Epithelial Permeability. Int. J. Mol. Sci. 2020, 21, 6402. [Google Scholar] [CrossRef]
- Pongkorpsakol, P.; Satianrapapong, W.; Wongkrasant, P.; Steinhagen, P.R.; Tuangkijkul, N.; Pathomthongtaweechai, N.; Muanprasat, C. Establishment of Intestinal Epithelial Cell Monolayers and Their Use in Calcium Switch Assay for Assessment of Intestinal Tight Junction Assembly. Methods Mol. Biol. 2021, 2367, 273–290. [Google Scholar] [CrossRef]
- Wongkrasant, P.; Pongkorpsakol, P.; Ariyadamrongkwan, J.; Meesomboon, R.; Satitsri, S.; Pichyangkura, R.; Barrett, K.E.; Muanprasat, C. A prebiotic fructo-oligosaccharide promotes tight junction assembly in intestinal epithelial cells via an AMPK-dependent pathway. Biomed. Pharmacother. 2020, 129, 110415. [Google Scholar] [CrossRef]
- Olivier, S.; Leclerc, J.; Grenier, A.; Foretz, M.; Tamburini, J.; Viollet, B. AMPK activation promotes tight junction assembly in intestinal epithelial caco-2 cells. Int. J. Mol. Sci. 2019, 20, 5171. [Google Scholar] [CrossRef]
- Krivan, H.C.; Clark, G.F.; Smith, D.F.; Wilkins, T.D. Cell surface binding site for Clostridium difficile enterotoxin: Evidence for a glycoconjugate containing the sequence Ga1α1-3Galβ1-4GlcNAc. Infect. Immun. 1986, 53, 573–581. [Google Scholar] [CrossRef]
- Rolfe, R.D.; Song, W. Purification of a functional receptor for Clostridium difficile toxin a from intestinal brush border membranes of infant hamsters. Clin. Infect. Dis. 1993, 16, S219–S227. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.D.; Wilkins, T.D. Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 1991, 59, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Claus, D. A standardized Gram staining procedure. World J. Microbiol. Biotechnol. 1992, 8, 451–452. [Google Scholar] [CrossRef]
- Wieser, A.; Schneider, L.; Jung, J.; Schubert, S. MALDI-TOF MS in microbiological diagnostics-identification of microorganisms and beyond (mini review). Appl. Microbiol. Biotechnol. 2012, 93, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Xu, J.; Zhou, H.; Dong, R.; Kang, M.; Zhao, J. Chitin oligosaccharide (COS) reduces antibiotics dose and prevents antibiotics-caused side effects in Adolescent Idiopathic Scoliosis (AIS) patients with spinal fusion surgery. Mar. Drugs 2017, 15, 70. [Google Scholar] [CrossRef]
- Pinna, A.; Donadu, M.G.; Usai, D.; Dore, S.; D’Amico-Ricci, G.; Boscia, F.; Zanetti, S. In vitro antimicrobial activity of a new ophthalmic solution containing povidone-iodine 0.6% (IODIM®). Acta Ophthalmol. 2020, 98, e178–e180. [Google Scholar] [CrossRef]
- Kothary, V.; Doster, R.S.; Rogers, L.M.; Kirk, L.A.; Boyd, K.L.; Romano-Keeler, J.; Haley, K.P.; Manning, S.D.; Aronoff, D.M.; Gaddy, J.A. Group B Streptococcus induces neutrophil recruitment to gestational tissues and elaboration of extracellular traps and nutritional immunity. Front. Cell. Infect. Microbiol. 2017, 7, 19. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavrogeni, M.E.; Asadpoor, M.; Judernatz, J.H.; van Ark, I.; Wösten, M.M.S.M.; Strijbis, K.; Pieters, R.J.; Folkerts, G.; Braber, S. Protective Effects of Alginate and Chitosan Oligosaccharides against Clostridioides difficile Bacteria and Toxin. Toxins 2023, 15, 586. https://doi.org/10.3390/toxins15100586
Mavrogeni ME, Asadpoor M, Judernatz JH, van Ark I, Wösten MMSM, Strijbis K, Pieters RJ, Folkerts G, Braber S. Protective Effects of Alginate and Chitosan Oligosaccharides against Clostridioides difficile Bacteria and Toxin. Toxins. 2023; 15(10):586. https://doi.org/10.3390/toxins15100586
Chicago/Turabian StyleMavrogeni, Maria Eleni, Mostafa Asadpoor, Jo H. Judernatz, Ingrid van Ark, Marc M. S. M. Wösten, Karin Strijbis, Roland J. Pieters, Gert Folkerts, and Saskia Braber. 2023. "Protective Effects of Alginate and Chitosan Oligosaccharides against Clostridioides difficile Bacteria and Toxin" Toxins 15, no. 10: 586. https://doi.org/10.3390/toxins15100586