Improved Production of Recombinant Carboxylesterase FumDM by Co-Expressing Molecular Chaperones in Pichia pastoris

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Construction and Selection of Recombinant P. pastoris Expressing Carboxylesterase FumDO/FumDM
2.2. Construction and Selection of Recombinant P. pastoris Co-Expressing Carboxylesterase FumDM and Molecular Chaperones
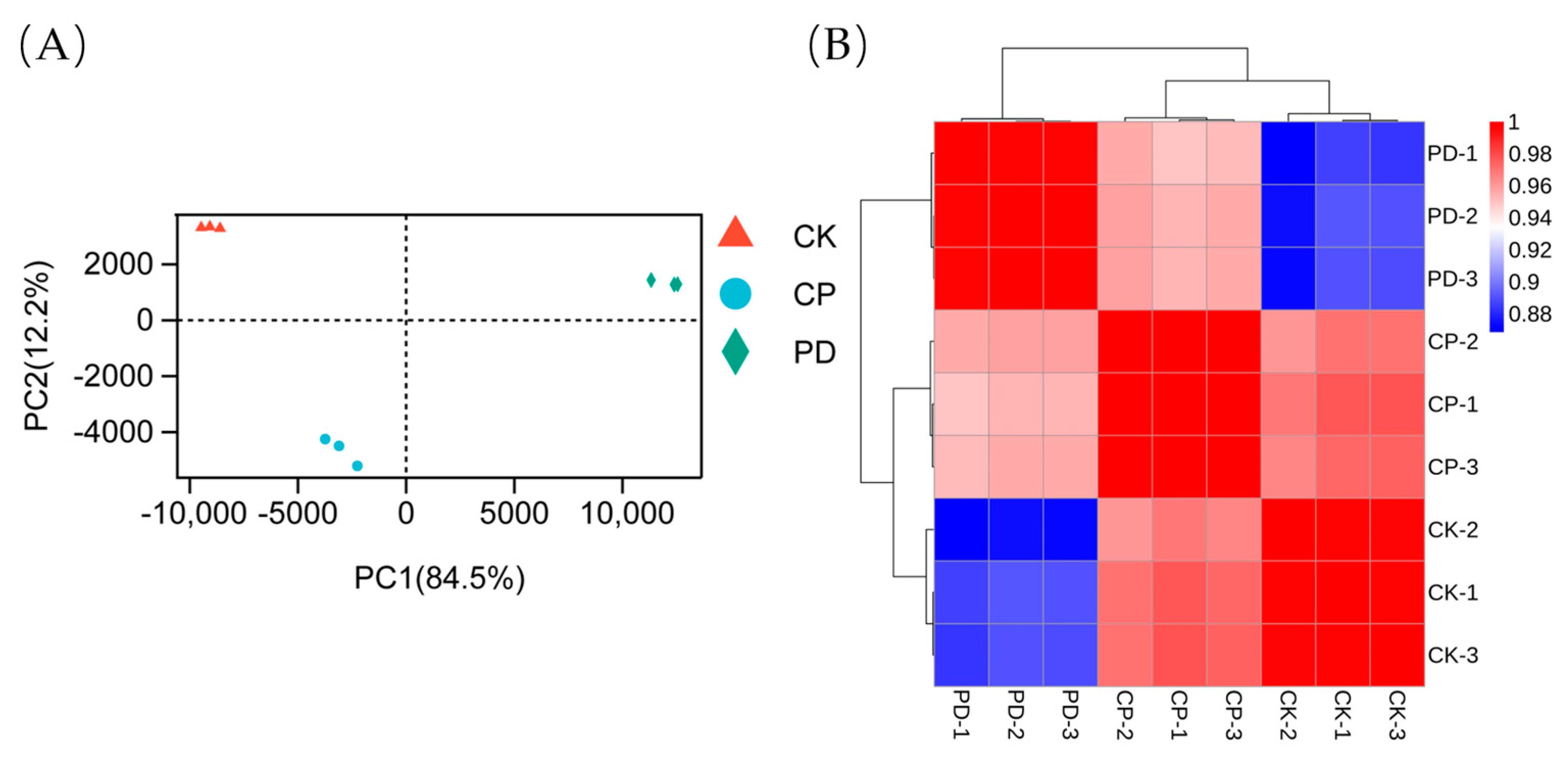
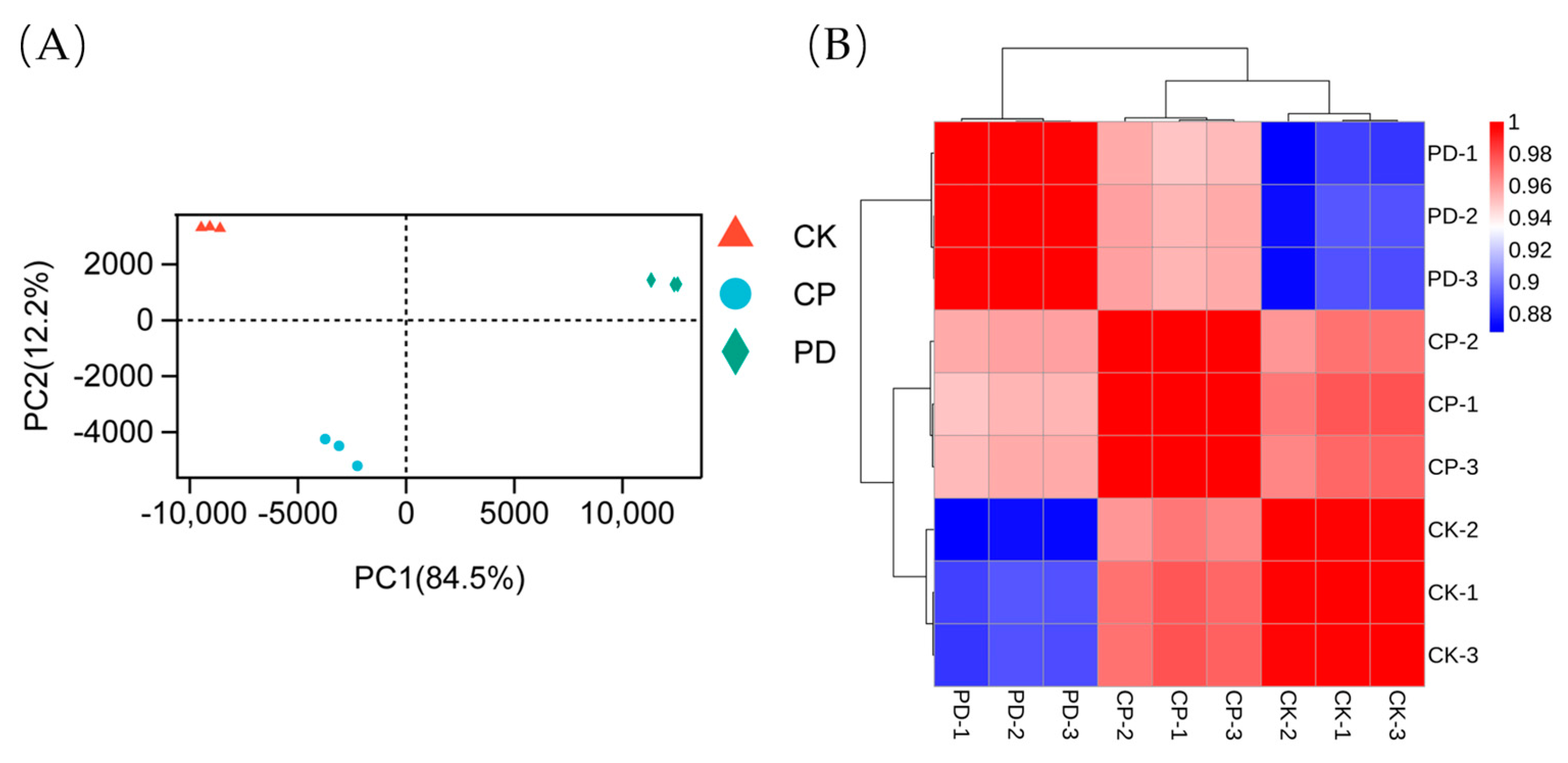
2.3. Transcriptomic Analysis
2.3.1. Sequencing, Evaluation, and Annotation of the Transcriptomic Profile
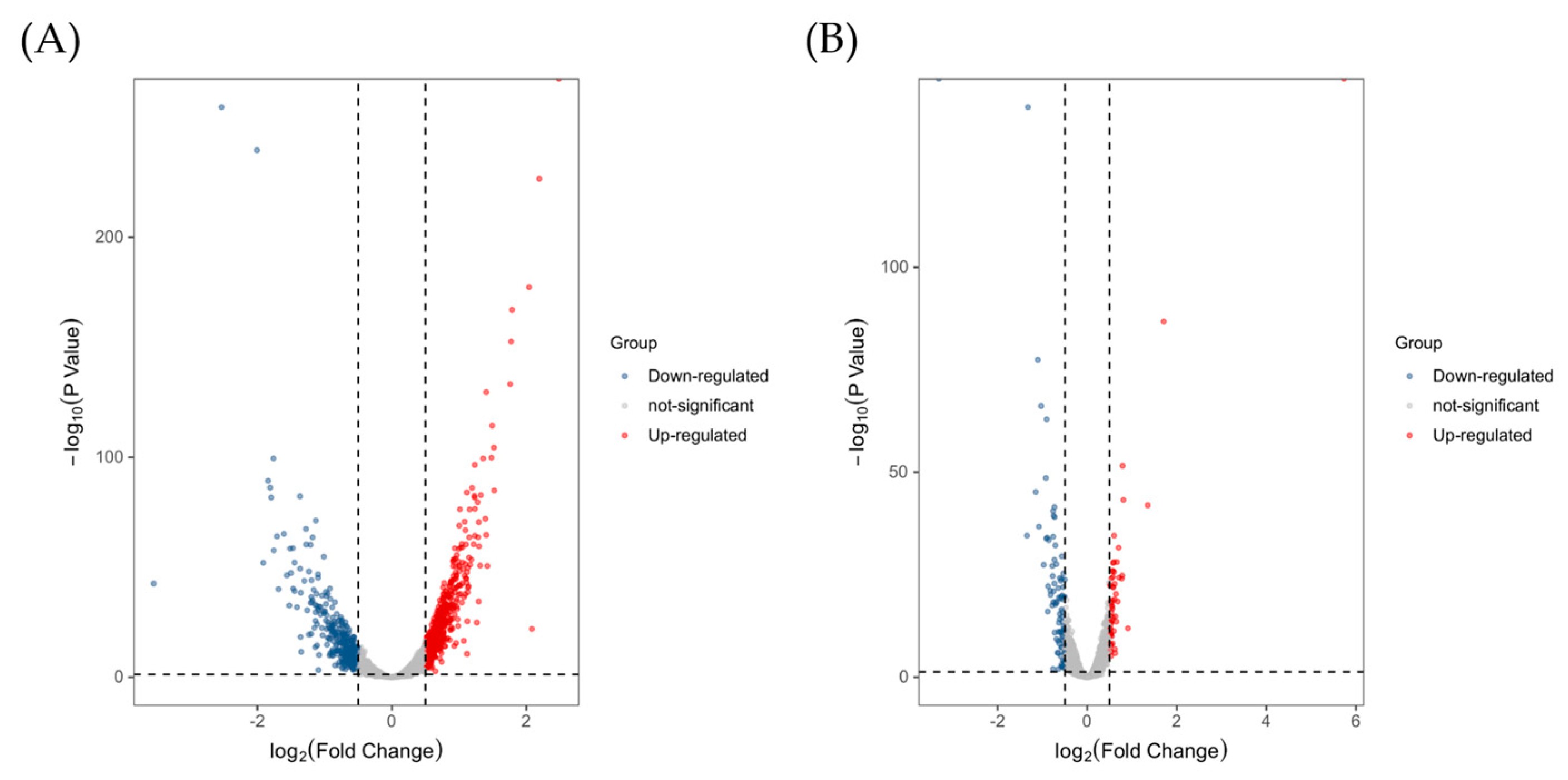
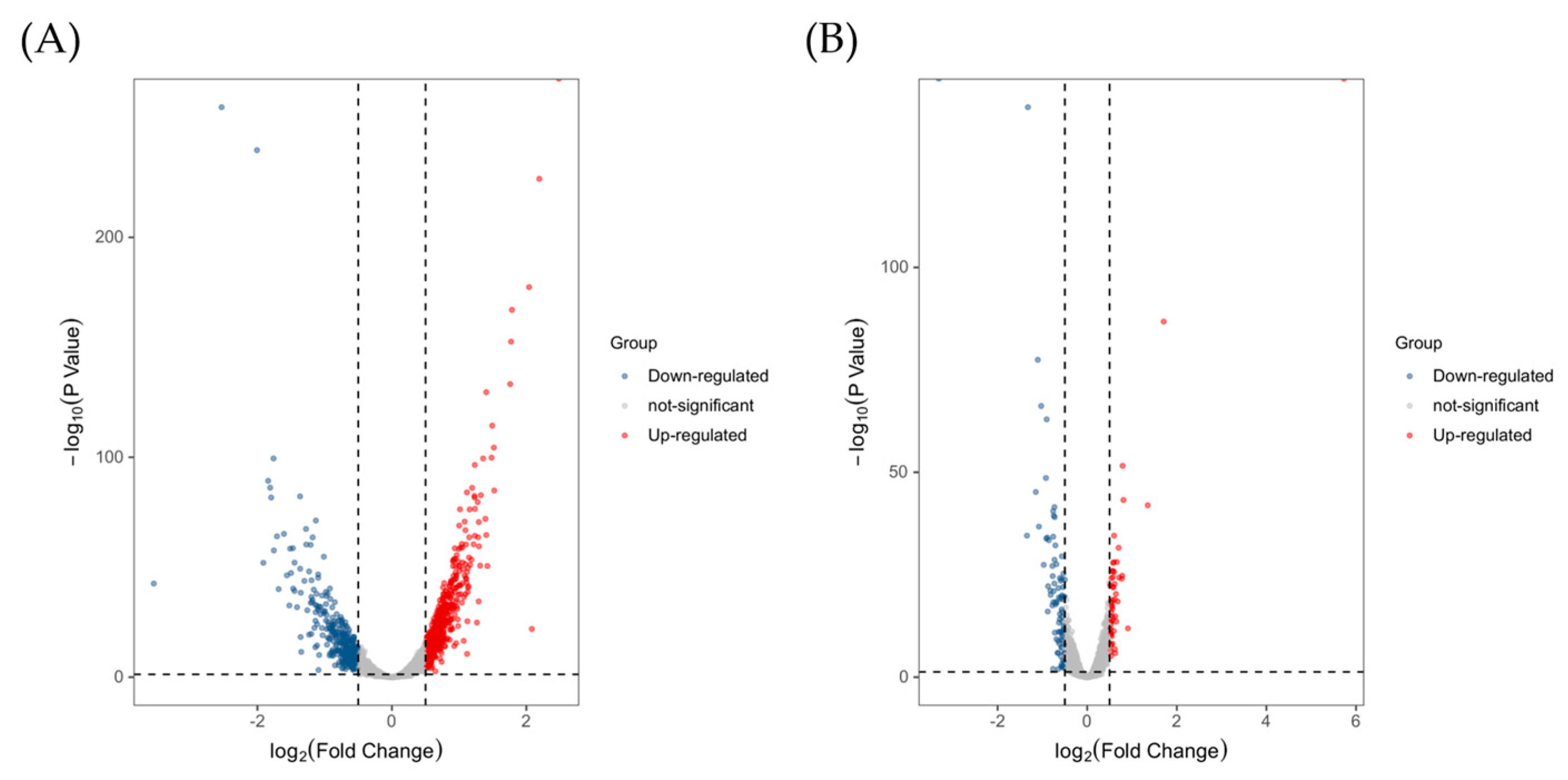
2.3.2. Functional Classification of DEGs and Pathway Analysis
2.3.3. Validation of DEGs by qRT-PCR
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Strains, Plasmids, and Medium
5.2. Construction of Recombinant Strains
5.3. Screening and Cultivation of Strains
5.4. Analysis of Protein Expression
5.5. Enzymatic Activity Assay by High Performance Liquid Chromatography
5.6. Transcriptome Analysis
5.7. Quantitative Real-Time PCR (qRT-PCR) Verification
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, C.; Song, G.; Lim, W. Effects of mycotoxin-contaminated feed on farm animals. J. Hazard. Mater. 2020, 389, 122087. [Google Scholar] [CrossRef] [PubMed]
- WHO. Mycotoxins. Available online: https://www.who.int/news-room/fact-sheets/detail/mycotoxins (accessed on 9 May 2018).
- Rheeder, J.P.; Marasas, W.; Vismer, H.F. Production of Fumonisin Analogs by Fusarium Species. Appl. Environ. Microbiol. 2002, 68, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Garcia, N.; Serna-Saldivar, S.O.; Garcia-Lara, S. Fumonisins and their analogues in contaminated corn and its processed foods–a review. Food Addit. Contam. 2018, 35, 2183–2203. [Google Scholar] [CrossRef] [PubMed]
- Odjo, S.; Alakonya, A.E.; Rosales-Nolasco, A.; Molina, A.L.; Muñoz, C.; Palacios-Rojas, N. Occurrence and postharvest strategies to help mitigate aflatoxins and fumonisins in maize and their co-exposure to consumers in Mexico and Central America. Food Control. 2022, 138, 108968. [Google Scholar] [CrossRef]
- Cao, C.; Zhu, X.; Li, X.; Ouyang, H.; Wang, K.; Li, X. Assessment of ionic homeostasis imbalance and cytochrome P450 system disturbance in mice during fumonisin B1 (FB1) exposure. Chemosphere 2020, 251, 126393. [Google Scholar] [CrossRef]
- Seo, E.; Yoon, Y.; Kim, K.; Shim, W.; Kuzmina, N.; Oh, K.; Lee, J.; Kim, D.; Suh, J.; Lee, S.; et al. Fumonisins B1 and B2 in agricultural products consumed in South Korea: An exposure assessment. J. Food Prot. 2009, 72, 436–440. [Google Scholar] [CrossRef]
- Wild, C.P.; Gong, Y.Y. Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 2010, 31, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Alsulami, T.; Nath, N.; Flemming, R.; Wang, H.; Zhou, W.; Yu, J.H. Development of a novel homogeneous immunoassay using the engineered luminescent enzyme NanoLuc for the quantification of the mycotoxin fumonisin B1. Biosens. Bioelectron. 2020, 177, 112939. [Google Scholar] [CrossRef]
- Xu, H.; Wang, L.; Sun, J.; Wang, L.; Guo, H.; Ye, Y.; Sun, X. Microbial detoxification of mycotoxins in food and feed. Crit. Rev. Food Sci. Nutr. 2021, 62, 11–19. [Google Scholar] [CrossRef]
- Heinl, S.; Hartinger, D.; Thamhesl, M.; Vekiru, E.; Krska, R.; Schatzmayr, G.; Moll, W.; Grabherr, R. Degradation of fumonisin B1 by the consecutive action of two bacterial enzymes. J. Biotechnol. 2010, 145, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; Liu, Z.; Jin, S.; Pan, K.; Liu, H.; Liu, T.; Li, X.; Zhang, C.; Luo, X.; et al. Biological detoxification of fumonisin by a novel carboxylesterase from Sphingomonadales bacterium and its biochemical characterization. Int. J. Biol. Macromol. 2021, 169, 18–27. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Additives and Products or Substances used in Animal Feed (FEEDAP). Scientific opinion on the safety and efficacy of fumonisin esterase (FUMzyme®) as a technological feed additive for pigs. EFSA J. 2014, 12, 3667–3686. [Google Scholar] [CrossRef]
- Duan, X.; Jiang, Z.; Liu, Y.; Yan, Q.; Xiang, M.; Yang, S. High-level expression of codon-optimized Thielavia terrestris cutinase suitable for ester biosynthesis and biodegradation. Int. J. Biol. Macromol. 2019, 135, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Jiwon, H.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Q.; Chen, L.; Zhang, C.; Bu, W.; Zhang, X.; Zhang, K.; Yang, Z. Improved production of recombinant Rhizomucor miehei lipase by coexpressing protein folding chaperones in Pichia pastoris, which triggered ER stress. Bioengineered 2020, 11, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Buchner, J. Molecular chaperones and protein quality control: An introduction to the JBC Reviews thematic series. J. Biol. Chem. 2019, 294, 2074–2075. [Google Scholar] [CrossRef]
- Freedman, R.B.; Klappa, P.; Ruddock, L.W. Model peptide substrates and ligands in analysis of action of mammalian protein disulfide-isomerase. Methods Enzymol. 2002, 348, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.G.; Hu, S.; Bankhead, A.; Neamati, N. Role of the ERO1-PDI interaction in oxidative protein folding and disease. Pharmacol. Ther. 2020, 210, 107525. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, L.; Heads, R.; Green, C.J. Cyclophilins and their possible role in the stress response. J. Exp. Pathol. 1999, 80, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G. Peptidyl-Prolyl cis-trans Isomerases and Their Effectors. Angew. Chem. Int. Ed. 1994, 33, 1415–1436. [Google Scholar] [CrossRef]
- Hampton, R.Y. ER stress response: Getting the UPR hand on misfolded proteins. Curr. Biol. 2000, 10, 518–521. [Google Scholar] [CrossRef]
- Qiang, L.; Wang, H.; Farmer, S.R. Adiponectin secretion is regulated by SIRT1 and the endoplasmic reticulum oxidoreductase Ero1-L alpha. Mol. Cell. Biol. 2007, 27, 4698–4707. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Woo, J.H.; Neville, D.M. Overexpression of an anti-CD3 immunotoxin increases expression and secretion of molecular chaperone BiP/Kar2p by Pichia pastoris. Appl. Environ. Microbiol. 2005, 71, 5332–5340. [Google Scholar] [CrossRef]
- Sharma, M.; Khurana, H.; Singh, D.N.; Negi, R. The genus Sphingopyxis: Systematics, ecology, and bioremediation potential—A review. J. Environ. Manag. 2020, 280, 111744. [Google Scholar] [CrossRef]
- Zheng, Y.; Lan, W.; Qiao, C.; Mulchandani, A.; Chen, W. Decontamination of vegetables sprayed with organophosphate pesticides by organophosphorus hydrolase and carboxylesterase (B1). Appl. Biochem. Biotechnol. 2007, 136, 233–241. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Z.; Zhang, T.; Wang, Y.; Yang, B. High-level expression of Thermomyces dupontii thermophilic lipase in Pichia pastoris via combined strategies. E3 J. Biotechnol. Pharm. Res. 2019, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Arevalo-Rodriguez, M.; Wu, X.; Hanes, S.D.; Heitman, J. Prolyl isomerases in yeast. Front. Biosci. Sch. Ed. 2004, 9, 2420–2446. [Google Scholar] [CrossRef] [PubMed]
- Weisshaar, N.; Welsch, H.; Guerra-Moreno, A.; Hanna, J.; Tansey, W. Phospholipase Lpl1 Links Lipid Droplet Function with Quality Control Protein Degradation. Mol. Biol. Cell. 2017, 28, 716–725. [Google Scholar] [CrossRef]
- Li, J.; Cai, J.; Ma, M.; Li, L.; Lu, L.; Wang, Y.; Wang, C.; Yang, J.; Xu, Z.; Yao, M.; et al. Preparation of a Bombyx mori acetylcholinesterase enzyme reagent through chaperone protein disulfide isomerase co-expression strategy in Pichia pastoris for detection of pesticides. Enzyme Microb. Technol. 2020, 144, 109741. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zhao, X.; Song, W.; He, N.; Jiang, S.; Zhou, Y.; Zhang, G. Combined strategies to improve the expression of acidic mammalian chitinase in Pichia pastoris for the production of N, N′-diacetylchitobiose. Biochem. Eng. J. 2021, 167, 107907. [Google Scholar] [CrossRef]
- Gething, M.J. Protein folding. The difference with prokaryotes. Nature 1997, 388, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Noi, K.; Inaba, K. Visualization of structural dynamics of protein disulfide isomerase enzymes in catalysis of oxidative folding and reductive unfolding. Curr. Opin. Struct. Biol. 2021, 66, 49–57. [Google Scholar] [CrossRef]
- Stamnes, M.A.; Rutherford, S.L.; Zuker, C.S. Cyclophilins: A new family of proteins involved in intracellular folding. Trends Cell Biol. 1992, 2, 272–276. [Google Scholar] [CrossRef]
- Zondlo, N.J. Aromatic-proline interactions: Electronically tunable CH/π interactions. Acc. Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef]
- Wu, W.J.; Raleigh, D.P. Local control of peptide conformation: Stabilization of cis proline peptide bonds by aromatic proline interactions. Biopolymers 1998, 45, 381–394. [Google Scholar] [CrossRef]
- Quon, E.; Sere, Y.Y.; Chauhan, N.; Johansen, J.; Sullivan, D.P.; Dittman, J.S.; Rice, W.J.; Chan, R.B.; Di, P.G.; Beh, C.; et al. Endoplasmic reticulum-plasma membrane contact sites integrate sterol and phospholipid regulation. PLoS Biol. 2018, 16, 2003864. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, J.; Wang, M.; Du, G.; Chen, J. Lactobacillus casei combats acid stress by maintaining cell membrane functionality. J. Ind. Microbiol. Biotechnol. 2012, 39, 1031–1039. [Google Scholar] [CrossRef]
- Görner, W.; Durchschlag, E.; Martinez-Pastor, M.T.; Estruch, F.; Ammerer, G.; Hamilton, B.; Ruis, H.; Schüller, C. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 1998, 12, 586–597. [Google Scholar] [CrossRef]
- Hong, S.Y.; Roze, L.V.; Wee, J.; Linz, J.E. Evidence that a transcription factor regulatory network coordinates oxidative stress response and secondary metabolism in aspergilli. MicrobiologyOpen 2013, 2, 144–160. [Google Scholar] [CrossRef]
- Jun, L.; Arne, H. The thioredoxin antioxidant system. Adv. Free Radical Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Montibus, M.; Pinson-Gadais, L.; Richard-Forget, F.; Barreau, C.; Ponts, N. Coupling of transcriptional response to oxidative stress and secondary metabolism regulation in filamentous fungi. Crit. Rev. Microbiol. 2015, 41, 295–308. [Google Scholar] [CrossRef]
- Enserink, J.M.; Hombauer, H.; Huang, M.; Kolodner, R.D. Cdc28/Cdk1 positively and negatively affects genome stability in S. cerevisiae. Int. J. Biochem. Cell Biol. 2009, 185, 423–437. [Google Scholar] [CrossRef]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational Regulation of Gene Expression during Conditions of Cell Stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Jürgen, B.; Hanschke, R.; Sarvas, M.; Hecker, M.; Schweder, T. Proteome and transcriptome based analysis of Bacillus subtilis cells overproducing an insoluble heterologous protein. Appl. Microbiol. Biotechnol. 2001, 55, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Liao, X. Function of Pichia pastoris ribosomal protein and regulation of exogenous protein synthesis. Ph.D. Thesis, South China University of Technology, Guangzhou, China, 2019. [Google Scholar] [CrossRef]
- Li, C.; Lin, Y.; Zheng, X.; Pang, N.; Liao, X.; Liu, X.; Huang, Y.; Liang, S. Combined strategies for improving expression of Citrobacter amalonaticus phytase in Pichia pastoris. BMC Biotechnol. 2015, 15, 88. [Google Scholar] [CrossRef] [PubMed]
- Steffen, K.K.; Mccormick, M.A.; Pham, K.M.; MacKay, V.L.; Delaney, J.R.; Murakami, C.J.; Kaeberlein, M.; Kennedy, B.K. Ribosome Deficiency Protects Against ER Stress in Saccharomyces cerevisiae. Genetics 2012, 191, 107–118. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains, Plasmids, and Primers | Description | References |
|---|---|---|
| Strains | ||
| E.coli DH5α | General cloning host | Takara |
| P. pastoris GS115 | Host strain (his4−, Mut+) | Invitrogen |
| GS115-FumDO | GS115 harboring pPIC9K-FumDO | This study |
| GS115-FumDM | GS115 harboring pPIC9K-FumDM | This study |
| GS115-FumDM/Bip | GS115-FumDM harboring pPICZA-Bip | This study |
| GS115-FumDM/HAC1 | GS115-FumDM harboring pPICZA-HAC1 | This study |
| GS115-FumDM/ERO1 | GS115-FumDM harboring pPICZA-ERO1 | This study |
| GS115-FumDM/CPR5 | GS115-FumDM harboring pPICZA-CPR5 | This study |
| GS115-FumDM/PDI | GS115-FumDM harboring pPICZA-PDI | This study |
| Plasmids | ||
| pPIC9K | Secretion-expression vector | Invitrogen |
| pPIC9K-FumDO | pPIC9K derivative, carrying FumDO gene expression cassette | This study |
| pPIC9K-FumDM | pPIC9K derivative, carrying FumDM gene expression cassette | This study |
| pPICZA | Intracellular expression vector | Invitrogen |
| pPICZA-Bip | pPICZA derivative, carrying Bip gene expression cassette | This study |
| pPICZA-HAC1 | pPICZA derivative, carrying HAC1 gene expression cassette | This study |
| pPICZA-ERO1 | pPICZA derivative, carrying ERO1 gene expression cassette | This study |
| pPICZA-CPR5 | pPICZA derivative, carrying CPR5 gene expression cassette | This study |
| pPICZA-PDI | pPICZA derivative, carrying PDI gene expression cassette | This study |
| Primers | ||
| FumDO-F | AGGCTGAAGCTTACGTAGAATTCATGGTGAAAGAGCACCAATGCCGTGG | FumDO amplification |
| FumDO-R | AAGGCGAATTAATTCGCGGCCGCCTATTTTGAGGGTTGGCAGGCTTTGC | FumDO amplification |
| FumDM-F | AGGCTGAAGCTTACGTAGAATTCATGGTTAAGGAACACCAATGTAGAGG | FumDM amplification |
| FumDM-R | AAGGCGAATTAATTCGCGGCCGCTTATTTAGAAGGTTGACAAGCCTTAG | FumDM amplification |
| Bip-F | ATTATTCGAAACGAGGAATTCGCCACCATGCTGTCGTTAAAACCATCTTG | Bip amplification |
| Bip-R | TGGCGGCCGCCGCGGCTCGAGGTCAACTCATCATGATCATAGTCATAG | Bip amplification |
| HAC1-F | CGGAATTCGCCACCATGCCCGTAGATTCTTCTCATAAG | HAC1 amplification |
| HAC1-R | CCGCTCGAGGTCCTGATCGCTATGCATGTCAAC | HAC1 amplification |
| ERO1-F | ATTATTCGAAACGAGGAATTCGCCACCATGAGGATAGTAAGGAGCGTAG | ERO1 amplification |
| ERO1-R | TGGCGGCCGCCGCGGCTCGAGGTCAAGTCTACTCTATATGTGGTATCTC | ERO1 amplification |
| CPR5-F | CGGAATTCGCCACCATGAAATTGTTGAACTTTCTGCTTAG | CPR5 amplification |
| CPR5-R | CCGCTCGAGGTCAACTCATCTTTCACGACCTC | CPR5 amplification |
| PDI-F | CGGAATTCGCCACCATGCAATTCAACTGGGATATTAAAAC | PDI amplification |
| PDI-R | CCGCTCGAGGTAAGCTCGTCGTGAGCGTCTG | PDI amplification |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Guan, X.; Liu, H.; Chang, X.; Sun, J.; Sun, C.; Zhao, C. Improved Production of Recombinant Carboxylesterase FumDM by Co-Expressing Molecular Chaperones in Pichia pastoris. Toxins 2023, 15, 156. https://doi.org/10.3390/toxins15020156
Jiang L, Guan X, Liu H, Chang X, Sun J, Sun C, Zhao C. Improved Production of Recombinant Carboxylesterase FumDM by Co-Expressing Molecular Chaperones in Pichia pastoris. Toxins. 2023; 15(2):156. https://doi.org/10.3390/toxins15020156
Chicago/Turabian StyleJiang, Lixiang, Xiao Guan, Hujun Liu, Xiaojiao Chang, Jing Sun, Changpo Sun, and Chengcheng Zhao. 2023. "Improved Production of Recombinant Carboxylesterase FumDM by Co-Expressing Molecular Chaperones in Pichia pastoris" Toxins 15, no. 2: 156. https://doi.org/10.3390/toxins15020156
APA StyleJiang, L., Guan, X., Liu, H., Chang, X., Sun, J., Sun, C., & Zhao, C. (2023). Improved Production of Recombinant Carboxylesterase FumDM by Co-Expressing Molecular Chaperones in Pichia pastoris. Toxins, 15(2), 156. https://doi.org/10.3390/toxins15020156