Comparative Aspects of Ricin Toxicity by Inhalation

Abstract
:1. Introduction
1.1. Ricin Toxin and Mechanism of Action
1.2. Respiratory Tract Deposition, Distribution and Clearance of Inhaled Ricin
1.3. Pathogenesis of Inhaled Ricin Toxicity
2. Animal Models of Ricin Toxicity by Inhalation
2.1. Toxicity of Inhaled Ricin

{kind=link}
| Strain | Exposure | LD50 (µg/kg) | Reference |
|---|---|---|---|
| Mouse | |||
| C57/BL6 | Intratracheal instillation | 10 (estimated) | [39] |
| BALB/c | Whole body | 14.0 | [30] |
| BALB/c | Whole body | 11.2 | Cited by [1] |
| BALB/c | Not stated | 1.0–10.0 (varying particle sizes) | [22] |
| BALB/c | Head only | 10.4 | [41] |
| BALB/c | Nose only | 0.58 (inconsistent aerosol concentration) | [14] |
| Swiss Webster | Nose only | 4.0 | [40] |
| BXSB | Not stated | 2.8 | Cited by [1] |
| CD-1 | Nose only | >3.0 | [42] |
| Rat | |||
| Porton Wistar | Head only | 3.7 (var. Hale Queen) | [1,45] |
| Porton Wistar | Head only | 9.8 (var. Zanzibariensis) | [1,45] |
| Sprague Dawley | Nose only | 0.24 (assuming 20% deposition and an assumed minute volume) | [14] |
| Rabbit | |||
| New Zealand White | Muzzle only | 3.0 | [37] |
| Pig | |||
| Yorkshire x Landrace | Intratracheal instillation | LD50 not stated. A 1 µg/kg does resulted in no signs, 3 µg/kg resulted in death of all animals. | [24] |
| Non-human primates | |||
| Rhesus macaque | Head only | 15.0 | Cited by [1] |
| Rhesus macaque | Head only | 15.5 | Dstl unpublished data |
| Rhesus macaque | Head only | 5.8 | [29] |
| African green monkey | Not stated | 5.8 | Cited by [1] |
2.2. Comparative Anatomy Relevant to Ricin Toxicity by Inhalation
2.3. Comparative Pathology of Acute Ricin Toxicity by Inhalation
2.3.1. Nasal Cavity
Rhesus Macaque
Rabbit (New Zealand White)
Rat (Sprague Dawley)
Mouse (BALB/c)
2.3.2. Larynx and Trachea
Rhesus Macaque
Rabbit (New Zealand White)
Rat (Porton Wistar)
Rat (Sprague Dawley)
Mouse (BALB/c)
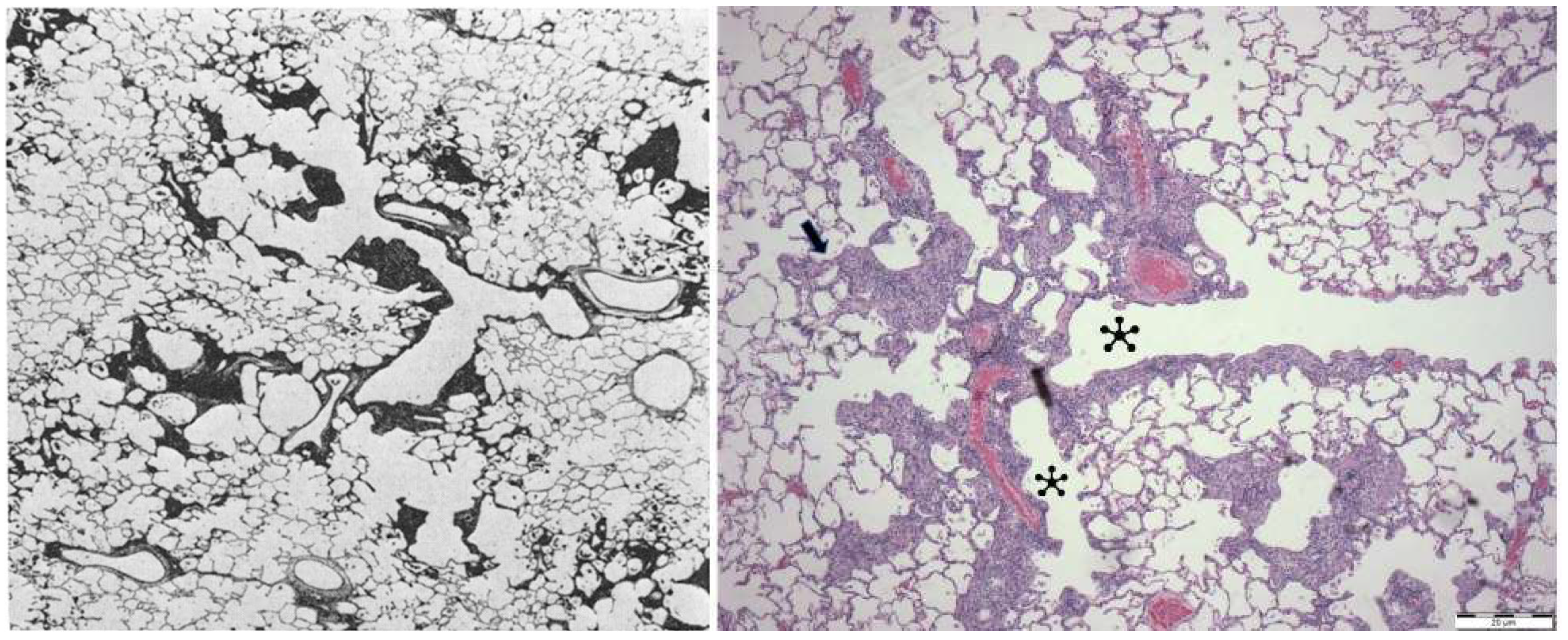
2.3.3. Lungs
Rhesus Macaque
Pig (Yorkshire x Landrace)
Rabbit (New Zealand White)
Rat (Porton Wistar)
Rat (Sprague Dawley)
Mouse (BALB/c)
Mouse (C57Bl/6)
2.3.4. Thoracic Cavity
Rhesus Macaque
Rabbit (New Zealand White)
2.3.5. Lymphoid Tissues
Rhesus Macaque
Rabbit (New Zealand White)
Rat (Porton Wistar)
Rat (Sprague Dawley)
Mouse (BALB/c)
2.4. Comparative Chronic Pathology of Ricin Toxicity by Inhalation
2.4.1. Nasal Cavity
Rhesus Macaque
Rabbit (New Zealand White, following Vaccination)
2.4.2. Larynx and Trachea
Rhesus Macaque
Rabbit (New Zealand White, following Vaccination)
2.4.3. Lungs
Rhesus Macaque
Rabbit (New Zealand White, following Vaccination)
Rat (Porton Wistar)
Rat (Sprague Dawley)
Mouse (BALB/c)
Mouse (C57BL/6)
Mouse (Swiss Webster) (Vaccinated)
2.4.4. Thoracic Cavity
Rhesus Macaque
2.4.5. Lymphoid Tissues
Rhesus Macaque
Rabbit (New Zealand White, following Vaccination)
2.5. Comparative Aspects of Animal Models of Pulmonary Fibrosis
3. Distant Organ Damage following Ricin Toxicity by Inhalation
3.1. Rhesus Macaque
3.2. Rat (Porton Wistar)
3.3. Mouse Models
3.4. Topical Exposure
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffiths, G.D.; Phillips, G.J.; Holley, J. Inhalation toxicology of ricin preparations: Animal models, prophylactic and therapeutic approaches to protection. Inhal. Toxicol. 2007, 19, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Falach, R.; Sapoznikov, A.; Gal, Y.; Israeli, O.; Leitner, M.; Seliger, N.; Ehrlich, S.; Kronman, C.; Sabo, T. Quantitative profiling of the in vivo enzymatic activity of ricin reveals disparate depurination of different pulmonary cell types. Toxicol. Lett. 2016, 258, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Simmons, B.M.; Stahl, P.D.; Russell, J.H. Mannose receptor-mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for a chain translocation. J. Biol. Chem. 1986, 261, 7912–7920. [Google Scholar] [CrossRef] [PubMed]
- Newton, D.L.; Wales, R.; Richardson, P.T.; Walbridge, S.; Saxena, S.K.; Ackerman, E.J.; Roberts, L.M.; Lord, J.M.; Youle, R.J. Cell surface and intracellular functions for ricin galactose binding. J. Biol. Chem. 1992, 267, 11917–11922. [Google Scholar] [CrossRef] [PubMed]
- Linehan, S.A.; Martínez-Pomares, L.; Stahl, P.D.; Gordon, S. Mannose receptor and its putative ligands in normal murine lymphoid and nonlymphoid organs: In situ expression of mannose receptor by selected macrophages, endothelial cells, perivascular microglia, and mesangial cells, but not dendritic cells. J. Exp. Med. 1999, 189, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Gal, Y.; Seliger, N.; Sabo, T.; Kronman, C. Diverse profiles of ricin-cell interactions in the lung following intranasal exposure to ricin. Toxins 2015, 7, 4817–4831. [Google Scholar] [CrossRef] [Green Version]
- Mooney, B.; Torres-Velez, F.J.; Doering, J.; Ehrbar, D.J.; Mantis, N.J. Sensitivity of Kupffer cells and liver sinusoidal endothelial cells to ricin toxin and ricin toxin-Ab complexes. J. Leukoc. Biol. 2019, 106, 1161–1176. [Google Scholar] [CrossRef]
- Schep, L.J.; Temple, W.A.; Butt, G.A.; Beasley, M.D. Ricin as a weapon of mass terror—Separating fact from fiction. Environ. Int. 2009, 35, 1267–1271. [Google Scholar] [CrossRef]
- Lindauer, M.; Wong, J.; Magun, B. Ricin Toxin Activates the NALP3 Inflammasome. Toxins 2010, 2, 1500–1514. [Google Scholar] [CrossRef] [Green Version]
- Polito, L.; Bortolotti, M.; Battelli, M.G.; Calafato, G.; Bolognesi, A. Ricin: An Ancient Story for a Timeless Plant Toxin. Toxins 2019, 11, 324. [Google Scholar] [CrossRef] [Green Version]
- David, J.; Wilkinson, L.J.; Griffiths, G.D. Inflammatory gene expression in response to sub-lethal ricin exposure in Balb/c mice. Toxicology 2009, 264, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Gal, Y.; Mazor, O.; Falach, R.; Sapoznikov, A.; Kronman, C.; Sabo, T. Treatments for Pulmonary Ricin Intoxication: Current Aspects and Future Prospects. Toxins 2017, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suntres, Z.E.; Stone, W.L.; Smith, M.G. Ricin-induced toxicity: The role of oxidative stress. J. Med. Chem. Biol. Radiol. Def. 2005, 3, 1–21. [Google Scholar]
- Benson, J.M.; Gomez, A.P.; Wolf, M.L.; Tibbetts, B.M.; March, T.H. The acute toxicity, tissue distribution, and histopathology of inhaled ricin in Sprague Dawley rats and BALB/c mice. Inhal. Toxicol. 2011, 23, 247–256. [Google Scholar] [CrossRef]
- Cook, D.L.; David, J.; Griffiths, G.D. Retrospective identification of ricin in animal tissues following administration by pulmonary and oral routes. Toxicology 2006, 223, 61–70. [Google Scholar] [CrossRef]
- Griffiths, G.D.; Knight, S.J.; Holley, J.; Thullier, P. Evaluation by ELISA of Ricin Concentration in Fluids and Tissues after Exposure to Aerosolised Ricin, and Evaluation of an Immunochromatographic Test for Field Diagnosis. J. Clin. Toxicol. 2013, 3, 1000162. [Google Scholar] [CrossRef] [Green Version]
- West, J.B.; Luks, A.M. West s Pulmonary Pathophysiology the Essentials, 9th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2017. [Google Scholar]
- West, J.B.; Luks, A.M. West’s Pulmonary Physiology the Essential, 10th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2016. [Google Scholar]
- Schlesinger, R.B. Comparative deposition of inhaled aerosols in experimental animals and humans: A review. J. Toxicol. Environ. Health 1985, 15, 197–214. [Google Scholar] [CrossRef]
- Darquenne, C. Deposition Mechanisms. J. Aerosol Med. Pulm. Drug Deliv. 2020, 33, 181–185. [Google Scholar] [CrossRef]
- Griffiths, G.D. Understanding ricin from a defensive viewpoint. Toxins 2011, 3, 1373–1392. [Google Scholar] [CrossRef] [Green Version]
- Roy, C.J.; Hale, M.; Hartings, J.M.; Pitt, L.; Duniho, S. Impact of inhalation exposure modality and particle size on the respiratory deposition of ricin in BALB/c mice. Inhal. Toxicol. 2003, 15, 619–638. [Google Scholar] [CrossRef]
- Lord, J.M.; Griffiths, G.D. Ricin: Chemistry, Sources, Exposures, Toxicology and Medical Aspects. In General, Applied and Systems Toxicology; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Katalan, S.; Falach, R.; Rosner, A.; Goldvaser, M.; Brosh-Nissimov, T.; Dvir, A.; Mizrachi, A.; Goren, O.; Cohen, B.; Gal, Y.; et al. A novel swine model of ricin-induced acute respiratory distress syndrome. Dis. Model. Mech. 2017, 10, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korcheva, V.; Wong, J.; Lindauer, M.; Jacoby, D.B.; Iordanov, M.S.; Magun, B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol. Immunol. 2007, 44, 2761–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindauer, M.L.; Wong, J.; Iwakura, Y.; Magun, B.E. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signaling. J. Immunol. 2009, 183, 1419–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Wang, Z.; Meng, S.; Zhao, Z.; Zhang, C.; Fu, Y.; Li, J.; Nie, X.; Zhang, C.; Liu, L.; et al. Effects of ricin on primary pulmonary alveolar macrophages. J. Int. Med. Res. 2019, 47, 3763–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapoznikov, A.; Gal, Y.; Falach, R.; Sagi, I.; Ehrlich, S.; Lerer, E.; Makovitzki, A.; Aloshin, A.; Kronman, C.; Sabo, T. Early disruption of the alveolar-capillary barrier in a ricin-induced ARDS mouse model: Neutrophil-dependent and -independent impairment of junction proteins. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L255–L268. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, M.; Didier, P.J.; Sivasubramani, S.K.; Doyle, L.A.; Holley, J.; Roy, C.J. Pathology of lethal and sublethal doses of aerosolized ricin in rhesus macaques. Toxicol. Pathol. 2014, 42, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DaSilva, L.; Cote, D.; Roy, C.; Martinez, M.; Duniho, S.; Pitt, M.L.; Downey, T.; Dertzbaugh, M. Pulmonary gene expression profiling of inhaled ricin. Toxicon 2003, 41, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar] [CrossRef]
- Wilhelmsen, C.L.; Pitt, M.L. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet. Pathol. 1996, 33, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Pincus, S.H.; Bhaskaran, M.; Brey, R.N., 3rd; Didier, P.J.; Doyle-Meyers, L.A.; Roy, C.J. Clinical and Pathological Findings Associated with Aerosol Exposure of Macaques to Ricin Toxin. Toxins 2015, 7, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Roy, C.J.; Brey, R.N.; Mantis, N.J.; Mapes, K.; Pop, I.V.; Pop, L.M.; Ruback, S.; Killeen, S.Z.; Doyle-Meyers, L.; Vinet-Oliphant, H.S.; et al. Thermostable ricin vaccine protects rhesus macaques against aerosolized ricin: Epitope-specific neutralizing antibodies correlate with protection. Proc. Natl. Acad. Sci. USA 2015, 112, 3782–3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, C.; Ehrbar, D.; Bohorova, N.; Bohorov, O.; Kim, D.; Pauly, M.; Whaley, K.; Rong, Y.; Torres-Velez, F.J.; Vitetta, E.S.; et al. A Humanized Monoclonal Antibody against the Enzymatic Subunit of Ricin Toxin Rescues Rhesus macaques from the Lethality of Aerosolized Ricin. bioRxiv 2018, 407817. [Google Scholar] [CrossRef]
- Roy, C.J.; Ehrbar, D.J.; Bohorova, N.; Bohorov, O.; Kim, D.; Pauly, M.; Whaley, K.; Rong, Y.H.; Torres-Velez, F.J.; Vitetta, E.S.; et al. Rescue of rhesus macaques from the lethality of aerosolized ricin toxin. JCI Insight 2019, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- McLain, D.E.; Lewis, B.S.; Chapman, J.L.; Wannemacher, R.W.; Lindsey, C.Y.; Smith, L.A. Protective effect of two recombinant ricin subunit vaccines in the New Zealand white rabbit subjected to a lethal aerosolized ricin challenge: Survival, immunological response, and histopathological findings. Toxicol. Sci. 2012, 126, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.F.; White, D.E. Ultrastructure of rat lung following inhalation of ricin aerosol. Int. J. Exp. Pathol. 1997, 78, 267–276. [Google Scholar] [CrossRef]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B. Intrapulmonary delivery of ricin at high dosage triggers a systemic inflammatory response and glomerular damage. Am. J. Pathol. 2007, 170, 1497–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallshaw, J.E.; Richardson, J.A.; Vitetta, E.S. RiVax, a recombinant ricin subunit vaccine, protects mice against ricin delivered by gavage or aerosol. Vaccine 2007, 25, 7459–7469. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, S.J.C.; Griffiths, G.D.; Jenner, D.C.; Gwyther, R.J.; Stahl, F.M.; Cork, L.J.; Holley, J.L.; Green, A.C.; Clark, G.C. Production, Characterisation and Testing of an Ovine Antitoxin against Ricin; Efficacy, Potency and Mechanisms of Action. Toxins 2017, 9, 329. [Google Scholar] [CrossRef] [Green Version]
- Doebler, J.A.; Wiltshire, N.D.; Mayer, T.W.; Estep, J.E.; Moeller, R.B.; Traub, R.K.; Broomfield, C.A.; Calamaio, C.A.; Thompson, W.L.; Pitt, M.L. The distribution of [125I]ricin in mice following aerosol inhalation exposure. Toxicology 1995, 98, 137–149. [Google Scholar] [CrossRef]
- Roy, C.J.; Song, K.; Sivasubramani, S.K.; Gardner, D.J.; Pincus, S.H. Animal models of ricin toxicosis. Curr. Top. Microbiol. Immunol. 2012, 357, 243–257. [Google Scholar]
- Worbs, S.; Köhler, K.; Pauly, D.; Avondet, M.A.; Schaer, M.; Dorner, M.B.; Dorner, B.G. Ricinus communis intoxications in human and veterinary medicine-a summary of real cases. Toxins 2011, 3, 1332–1372. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, G.D.; Rice, P.; Allenby, A.C.; Bailey, S.C.; Upshall, D.G. Inhalation toxicology and histopathology of ricin and abrin toxins. Inhal. Toxicol. 1995, 7, 269–288. [Google Scholar] [CrossRef]
- Haley, P.J. The lymphoid system: A review of species differences. J. Toxicol. Pathol. 2017, 30, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, P.J. Species differences in the structure and function of the immune system. Toxicology 2003, 188, 49–71. [Google Scholar] [CrossRef] [PubMed]
- Herbert, R.A.; Janarshan, K.S.; Pandiri, A.R.; Cesta, M.F.; Miller, R.A. Nose, larynx, and trachea. In Boorman’s Pathology of the Rat, 2nd ed.; Suttie, A., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: London, UK, 2018. [Google Scholar]
- Herbert, R.A.; Janardhan, K.S.; Pandiri, A.R.; Cesta, M.F.; Chen, V.; Miller, R.A. Lung, pleura, and mediastinum. In Boorman’s Pathology of the Rat, 2nd ed.; Suttie, A., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: London, UK, 2018. [Google Scholar]
- Suarez, C.J.; Dintzis, S.M.; Frevert, C.W. Respiratory. In Comparative Anatomy and Histology a Mouse and Human Atlas, 1st ed.; Treuting, P.M., Dintzis, S.M., Eds.; Academic Press: Cambridge, MA, USA; Elsevier: London, UK, 2012. [Google Scholar]
- Chamanza, R.; Wright, J.A. A Review of the Comparative Anatomy, Histology, Physiology and Pathology of the Nasal Cavity of Rats, Mice, Dogs and Non-human Primates. Relevance to Inhalation Toxicology and Human Health Risk Assessment. J. Comp. Pathol. 2015, 153, 287–314. [Google Scholar] [CrossRef]
- Booth, W.D.; Baldwin, B.A.; Poynder, T.M.; Bannister, L.H.; Gower, D.B. Degeneration and regeneration of the olfactory epithelium after olfactory bulb ablation in the pig: A morphological and electrophysiological study. Q. J. Exp. Physiol. 1981, 66, 533–540. [Google Scholar] [CrossRef]
- Lum, H.; Mitzner, W. A species comparison of alveolar size and surface forces. J. Appl. Physiol. 1987, 62, 1865–1871. [Google Scholar] [CrossRef]
- Ackerman, M. Respiratory Tract. In Biology of the Domestic Pig; Pond, W., Mersmann, H., Eds.; Cornell University Press: Ithaca, NY, USA, 2001; pp. 502–532. [Google Scholar]
- Krejci, J.; Nechvatalova, K.; Blahutkova, M.; Faldyna, M. The respiratory tract in pigs and its immune system: A review. Vet. Med. 2013, 58, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, L.F.; Saraiva, L.R.; Kuang, D.; Yoon, K.H.; Buck, L.B. Olfactory receptor patterning in a higher primate. J. Neurosci. 2014, 34, 12241–12252. [Google Scholar] [CrossRef] [Green Version]
- Judge, E.P.; Hughes, J.M.; Egan, J.J.; Maguire, M.; Molloy, E.L.; O’Dea, S. Anatomy and bronchoscopy of the porcine lung. A model for translational respiratory medicine. Am. J. Respir. Cell Mol. Biol. 2014, 51, 334–343. [Google Scholar] [CrossRef]
- Parent, R. Comparative Biology of the Normal Lung, 2nd ed.; Elsevier: London, UK, 2015. [Google Scholar]
- Choi, R.; Goldstein, B.J. Olfactory epithelium: Cells, clinical disorders, and insights from an adult stem cell niche. Laryngoscope Investig. Otolaryngol. 2018, 3, 35–42. [Google Scholar] [CrossRef]
- Jeffrey, A.M.; Iatropoulos, M.J.; Williams, G.M. Nasal cytotoxic and carcinogenic activities of systemically distributed organic chemicals. Toxicol. Pathol. 2006, 34, 827–852. [Google Scholar] [CrossRef] [PubMed]
- Strocchi, P.; Dozza, B.; Pecorella, I.; Fresina, M.; Campos, E.; Stirpe, F. Lesions caused by ricin applied to rabbit eyes. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1113–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ASKJPC Joint Pathology Center Systemic Pathology Respiratory System September 2017 P-T04. Available online: https://www.askjpc.org/vspo/show_page.php?id=aUtBWFpmVnh4S0NKbFNsN1d1bXMzUT09 (accessed on 23 February 2021).
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L152–L160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal models of fibrotic lung disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Heppleston, A.G.; Leopo Ld, J.G. Chronic pulmonary emphysema: Anatomy and pathogenesis. Am. J. Med. 1961, 31, 279–291. [Google Scholar] [CrossRef]
- Van Slyke, G.; Sully, E.K.; Bohorova, N.; Bohorov, O.; Kim, D.; Pauly, M.H.; Whaley, K.J.; Zeitlin, L.; Mantis, N.J. Humanized Monoclonal Antibody That Passively Protects Mice against Systemic and Intranasal Ricin Toxin Challenge. Clin. Vaccine Immunol. 2016, 23, 795–799. [Google Scholar] [CrossRef] [Green Version]
- Falach, R.; Sapoznikov, A.; Evgy, Y.; Aftalion, M.; Makovitzki, A.; Agami, A.; Mimran, A.; Lerer, E.; Ben David, A.; Zichel, R.; et al. Post-Exposure Anti-Ricin Treatment Protects Swine Against Lethal Systemic and Pulmonary Exposures. Toxins 2020, 12, 354. [Google Scholar] [CrossRef]
- Falach, R.; Sapoznikov, A.; Alcalay, R.; Aftalion, M.; Ehrlich, S.; Makovitzki, A.; Agami, A.; Mimran, A.; Rosner, A.; Sabo, T.; et al. Generation of Highly Efficient Equine-Derived Antibodies for Post-Exposure Treatment of Ricin Intoxications by Vaccination with Monomerized Ricin. Toxins 2018, 10, 466. [Google Scholar] [CrossRef] [Green Version]
- Pratt, T.S.; Pincus, S.H.; Hale, M.L.; Moreira, A.L.; Roy, C.J.; Tchou-Wong, K.M. Oropharyngeal aspiration of ricin as a lung challenge model for evaluation of the therapeutic index of antibodies against ricin A-chain for post-exposure treatment. Exp. Lung Res. 2007, 33, 459–481. [Google Scholar] [CrossRef]
- Phalen, R.F.; Mendez, L.B. Dosimetry considerations for animal aerosol inhalation studies. Biomarkers 2009, 14 (Suppl. 1), 63–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef] [PubMed]
| Anatomical Feature | Human | Rhesus Macaque | Pig | Rat | Mouse |
|---|---|---|---|---|---|
| Obligate nasal breather | No | No | No | Yes | Yes |
| Complex turbinates | No | No | Yes | Yes | Yes |
| Right lung Left lung | 3 lobes 2 lobes | 4 lobes 3 lobes | 4 lobes 3 lobes | 4 lobes 1 lobe | 4 lobes 1 lobe |
| Airway branching | Dichotomous branching | Monopodial branching | |||
| Nasal Cavity | |||||
| Vestibule | Stratified squamous epithelium (thinner in rodents). | ||||
| Nasal transitional epithelium | Located between stratified squamous epithelium and respiratory epithelium. | ||||
| Thick, stratified, few ciliated, epithelium, including occasional goblet cells. | Non-ciliated columnar epithelium. | Thin, pseudostratified, non-ciliated, basal, columnar and cuboidal epithelium with scattered ciliated cells. Contains abundant smooth endoplasmic reticulum (P450) for xenometabolism. | |||
| Nasal respiratory epithelium | Covers most of the nasal cavity of primates. Psuedostratified, ciliated, columnar epithelium. Gradual increase in goblet cell density anterior to posterior. Varies according to intranasal location. | Psuedostratified, ciliated, columnar epithelium with goblet cells appearing by the nasopharynx. Thick basal lamina. | <50% nasal cavity. Ciliated, goblet and basal cells. Lesser numbers of non-ciliated cells, chemoreceptor and brush cells. P450 for xenometabolism. Varies according to intranasal location. | ||
| Nasal olfactory epithelium | <3% of nasal cavity of human. Lines the medial superior vertical lamellae of the superior turbinates and the corresponding superior septum. There is, however, considerable variability among individuals. | ~5% of nasal cavity in monkeys. Present on dorsal parts of the nasal septum, medial turbinates, and lateral wall. | Present on caudodorsal, portion of nasal cavity, dorsal nasal meatus, nasal septum and ethmoturbinates. | ~50% of nasal cavity of rats and mice. P450 for xenometabolism. Lines upper caudal third of the septum, dorsal meatus, caudolateral wall and dorsal aspects of ethmoturbinates. | |
| Psuedostratified, columnar, neuroepithelium. 3 cell types: Olfactory sensory neurons, basal cells and sustentacular cells. | |||||
| Anatomical feature | Human | Rhesus macaque | Pig | Rat | Mouse |
| Lateral nasal gland | Absent | Secretory products drain into nasal vestibule. Major site of synthesis and secretion of odorant-binding proteins. Synthesises large amounts of immunoglobulin A. High metabolic capacity, potential target for secondary metabolites of inhaled or ingested xenobiotics. | |||
| Trachea | |||||
| Ciliated cells | 49% | 33% | Present | 32–41% | 39% |
| Club cells | 0% | 0% | Not reported | 0% | 49% |
| Goblet cells | 9% | 17% | Present | <1–2% | <1% |
| Serous cells | 0% | 0% | Not quantified | 27–42% | <1% |
| Basal cells | 33% | 42% | Not quantified | 13–27% | 10% |
| Other | 9% | 8% | Not quantified | 0–13% | 1% |
| Proximal intrapulmonary airways | |||||
| Ciliated cells | 37% | 44% | Present | 35% | 28–36% |
| Club cells | 0% | 0% | Not reported | 0% | 59–61% |
| Goblet cells | 10% | 15% | Present | <1% | <1% |
| Serous cells | 3% | 0% | Not quantified | 21% | <1% |
| Basal cells | 32% | 32% | Not quantified | 27% | <1% |
| Other | 18% | 8% | Not quantified | 16% | 2–14% |
| Terminal bronchioles | |||||
| Ciliated cells | 52% | 50% | Proximally only | 55% | 20–40% |
| Club cells | 0% | 0% | Present | 37%/40–65% | 60–80% |
| Goblet cells | Present | 20% | Absent | 0% | 0% |
| Serous cells | 35% | Not quantified | Not quantified | 0% | 0% |
| Basal cells | <1% | 10% | Not quantified | 0–<1% | <1% |
| Other | 13% | Not quantified | Not quantified | 8% | 0% |
| Respiratory bronchioles | |||||
| Ciliated cells | Present | <10% | Respiratory bronchioles often lacking (further characterisation of variation between strains required). | ||
| Club cells | Present | >90% | |||
| Goblet cells | Present | Present | |||
| Serous cells | Present | Present | |||
| Basal cells | Present | Present | |||
| Alveoli | |||||
| Size (mean linear intercept) | 210 µm | 200 µm | 133 µm | 100 µm | 80 µm |
| Blood–gas barrier thickness | 0.62 µm | 0.65 µm | 0.2–0.6 µm | Not reported | 0.32 µm |
| Airway mucus | |||||
| Mucus and mucin glycoproteins | Species differences and significances to be determined. Pigs have numerous submucosal glands in the trachea and bronchi. | ||||
| Reference | Countermeasure | Route of Entry | Dose | End Point(s) |
|---|---|---|---|---|
| Rhesus macaque | ||||
| [32] | None | Inhalation, head only | 27.2–41.8 µg/kg | 48 h |
| Dstl unpublished data | Antitoxin | Inhalation, head only | 3 × LD50 | 11–14, 90 days |
| [29] | None | Inhalation, head only | 13.2–27.3 µg/kg (lethal) 1.9–5.2 µg/kg (sublethal) | 30–48 h, 11 days |
| [33] | None | Inhalation, head only | 9.4–38.5 µg/kg (lethal) ~3 µg/kg (sublethal) | 25–52 h, 11–20 days |
| [34] | Vaccination | Inhalation, head only | 12–38 µg/kg | 14 days |
| [35,36] | Antitoxin (monoclonal antibody) 4 or 12 h after exposure | Inhalation, head only | 3 × LD50 | 14 days |
| Pig | ||||
| [24] | None | Intratracheal instillation | 3 µg/kg | 30–70 h |
| Rabbit | ||||
| [37] | Vaccination | Inhalation, muzzle only | 10–30 × LD50 | 95 days |
| [61] | None or topical wash | Topical ocular | 1 µg–100µg | 24 h, 7 days |
| Rat (Porton strain) | ||||
| Dstl unpublished data | None | Inhalation, head only | Various | 21 days |
| [38] | None | Inhalation | 11.21 mg/min/m3 (LCt30) | 48 h |
| Reference | Countermeasure | Route of entry | Dose | End point(s) |
| Rat (Sprague Dawley) | ||||
| [14] | None | Inhalation, nose only | 0.12 µg/L | 7 days |
| Mouse (BALB/c) | ||||
| [14] | None | Inhalation, nose only | 0.01 µg/L | 7 days |
| [30] | None | Inhalation, whole-body exposure | 14 µg/kg | 196 h |
| [11] | None | Inhalation, head only | 11.08 µg/kg | 96 h |
| Dstl unpublished data | None | Inhalation, head only | 3.5 or 5.2 mg/min/m3 | 96 days |
| Mouse (C57Bl/6) | ||||
| [39] | None | Intratracheal instillation | 20, 50 and 200 µg/kg | 48 h, 4 weeks |
| Swiss Webster | ||||
| [40] | Vaccination | Inhalation, nose only | 1–16 µg/kg (inhalation) | 14 days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© Crown copyright (2023), Dstl. This material is licensed under the terms of the Open Government Licence except where otherwise stated. To view this licence, visit http://www.nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Ar-chives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Share and Cite
Stoll, A.; Shenton, D.P.; Green, A.C.; Holley, J.L. Comparative Aspects of Ricin Toxicity by Inhalation. Toxins 2023, 15, 281. https://doi.org/10.3390/toxins15040281
Stoll A, Shenton DP, Green AC, Holley JL. Comparative Aspects of Ricin Toxicity by Inhalation. Toxins. 2023; 15(4):281. https://doi.org/10.3390/toxins15040281
Chicago/Turabian StyleStoll, Alexander, Daniel P. Shenton, A. Christopher Green, and Jane L. Holley. 2023. "Comparative Aspects of Ricin Toxicity by Inhalation" Toxins 15, no. 4: 281. https://doi.org/10.3390/toxins15040281
APA StyleStoll, A., Shenton, D. P., Green, A. C., & Holley, J. L. (2023). Comparative Aspects of Ricin Toxicity by Inhalation. Toxins, 15(4), 281. https://doi.org/10.3390/toxins15040281