Effects of C-Terminal Lys-Arg Residue of AapA1 Protein on Toxicity and Structural Mechanism


Abstract
:1. Introduction
2. Results
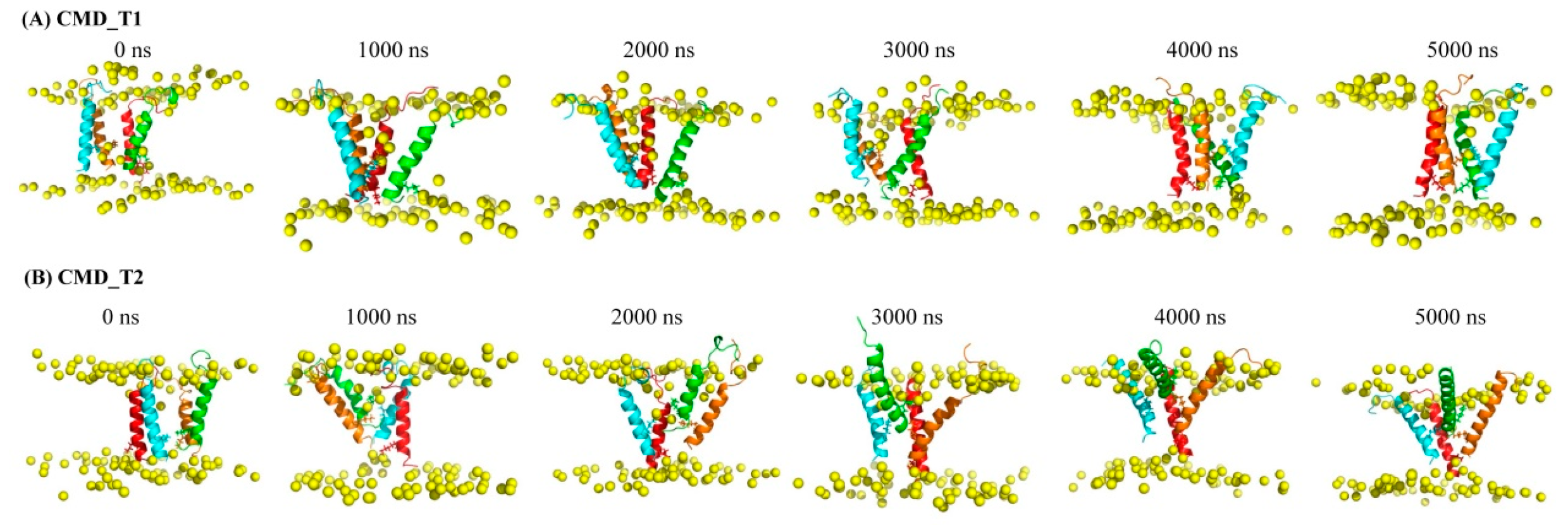
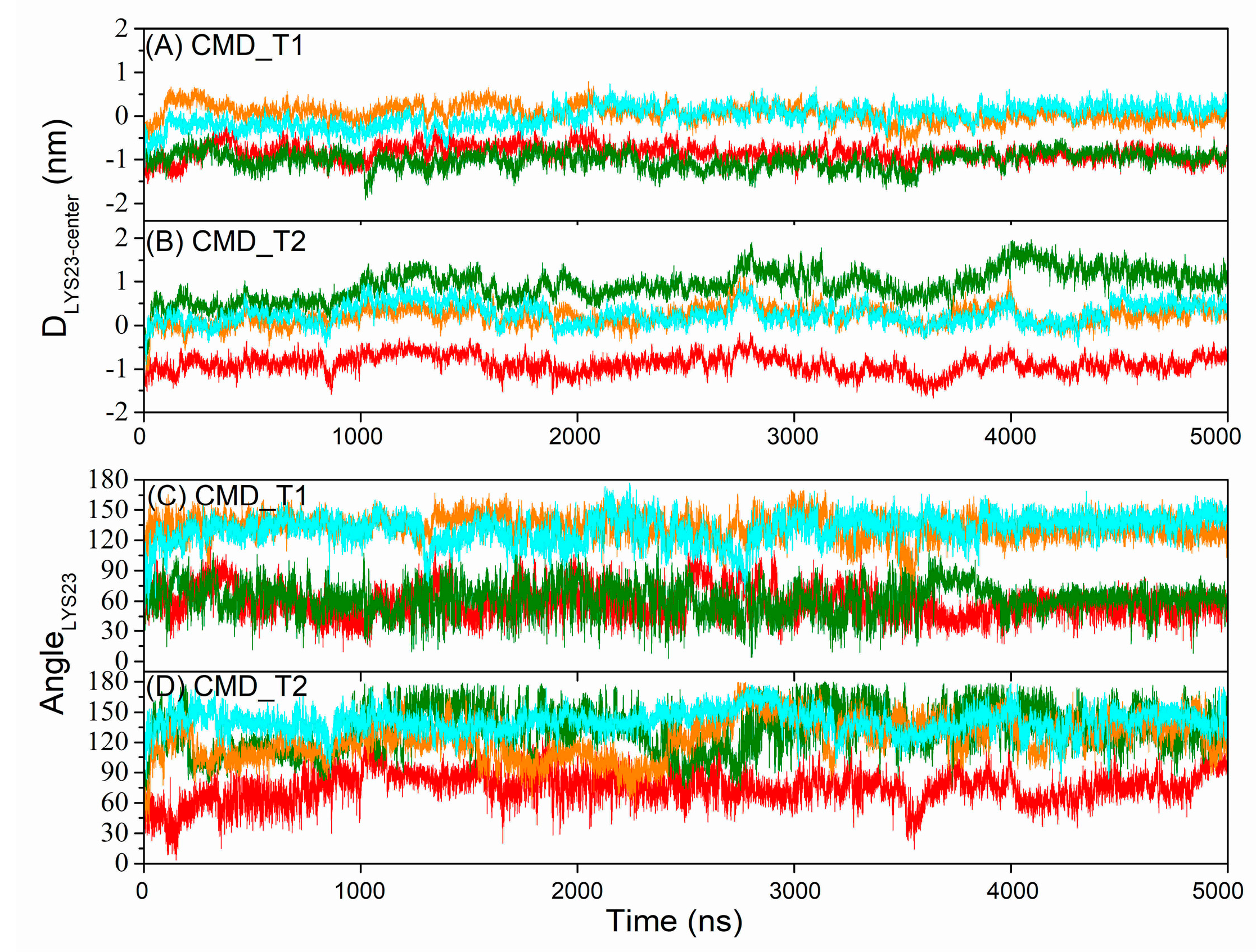
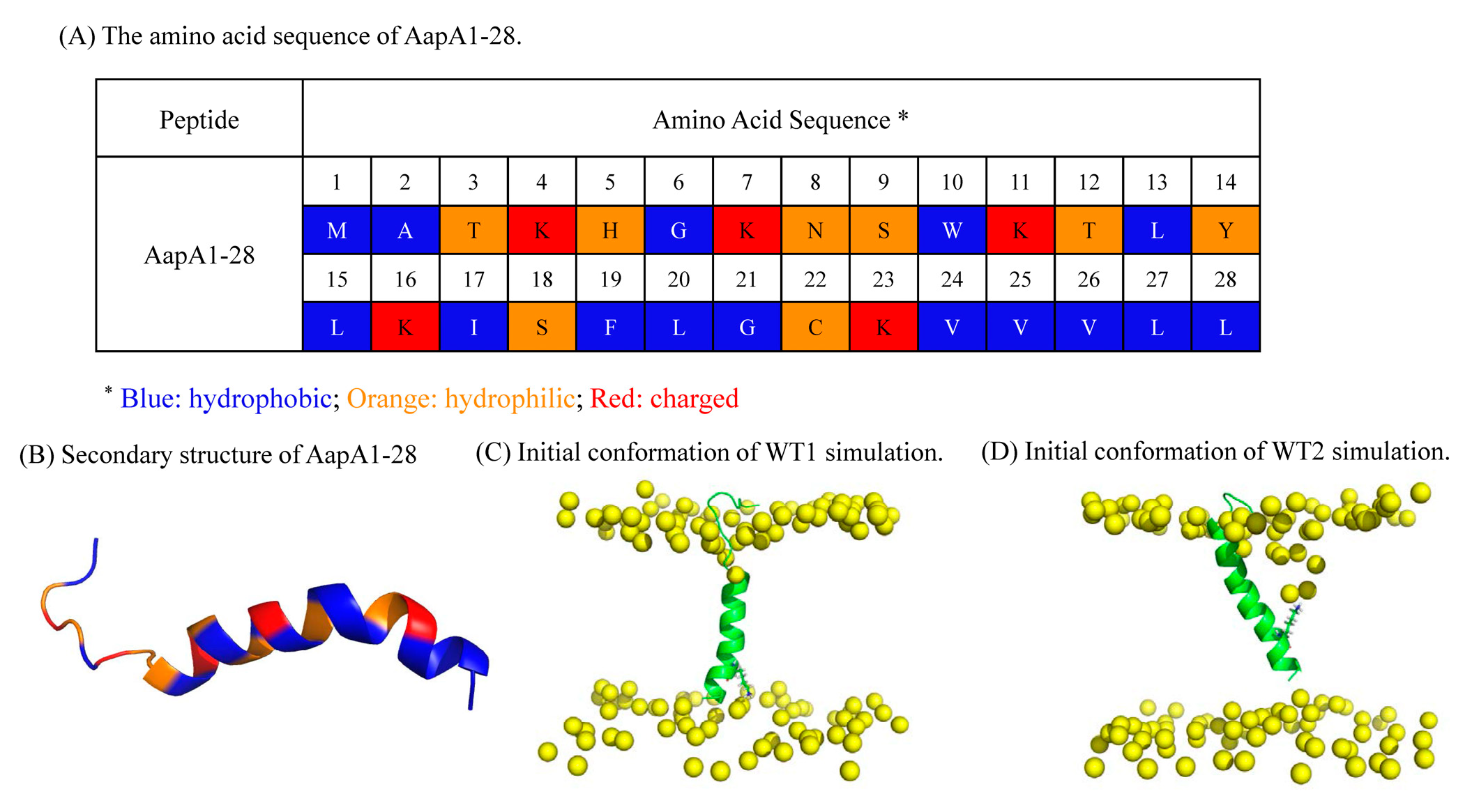
2.1. Structural and Positional Changes of Tetrameric AapA1-28 Protein Elucidated through CMD_T1/2 Simulations
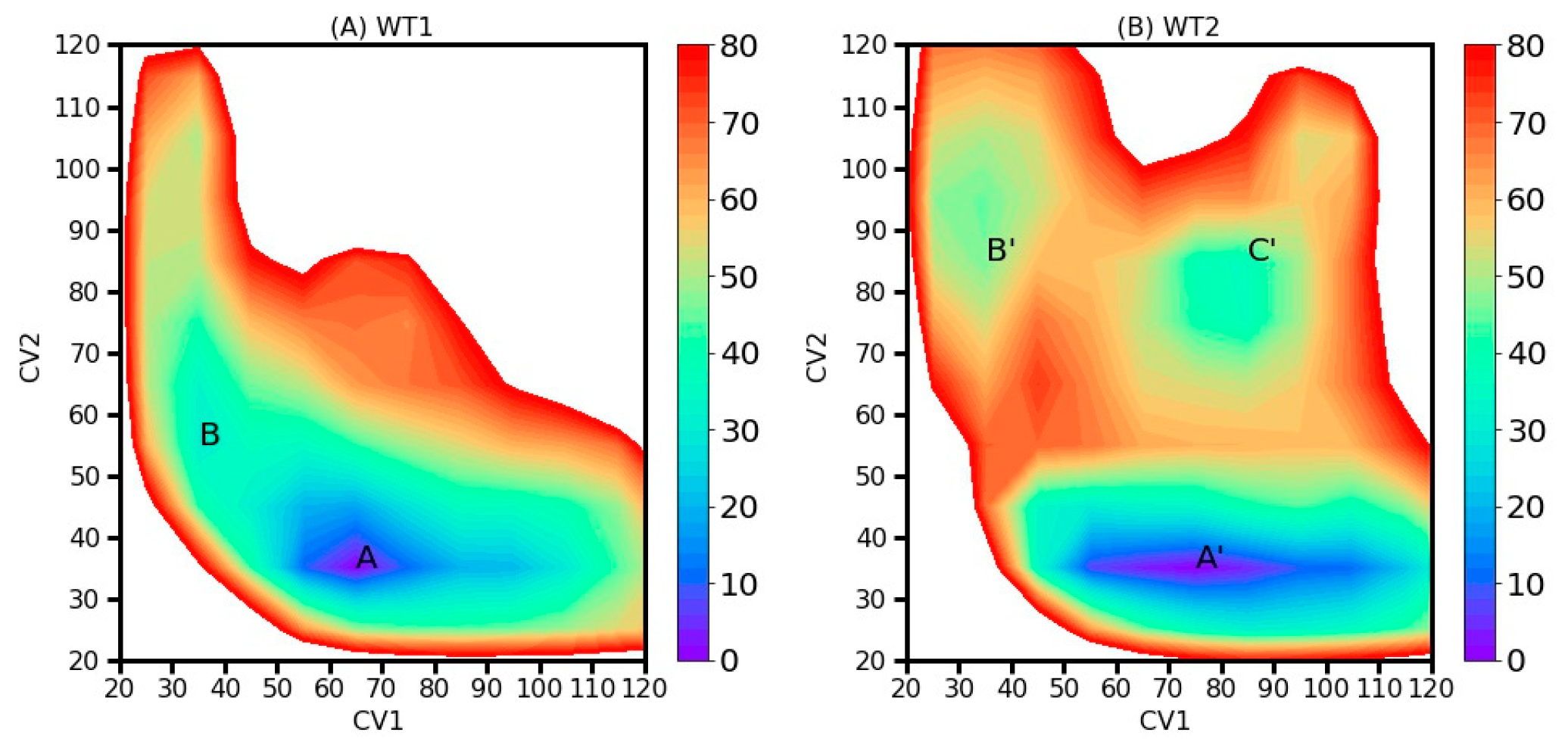
2.2. Two-Dimensional Free Energy Landscapes (FELs) from 2D-MetaD Simulations
2.3. Interactions between the AapA1-28 Protein and Membrane

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Simulation Systems | Sampling Method | The Initial Conformation | Two Collective Variables (CVs) | Acronym | Simulation Length (ns) |
|---|---|---|---|---|---|
| Tetramer AapA1-28/POPE/POPG (3:1) | CMD simulation | Figure 1A | CMD_T1 | 5000 ns | |
| Figure 1B | CMD_T2 | 5000 ns | |||
| AapA1-28/POPE/POPG (3:1) | 2D-MetaD simulation | Figure 3C | CV1: Contact number between K-23 and the top phosphate group. CV2: Contact number between K-23 and the bottom phosphate group. | WT1 | 2 replica× 1000 ns |
| AapA1-28/POPE/POPG (3:1) | 2D-MetaD simulation | Figure 3D | The same CVs | WT2 | 2 replica ×1000 ns |
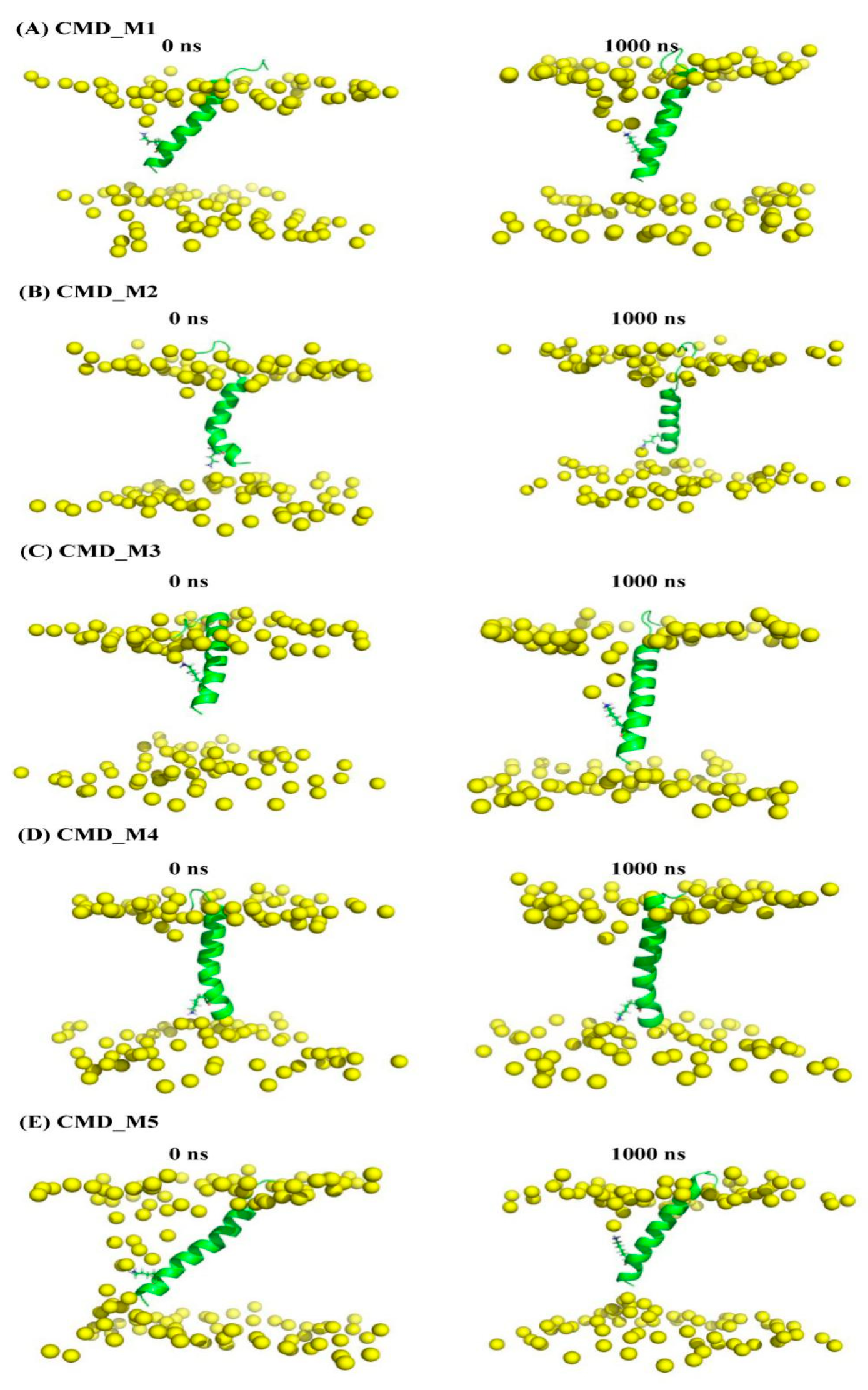
| AapA1-28/POPE/POPG (3:1) | CMD simulation | Figure 5A (representative conformation of region A in Figure 4) | CMD_M1 | 1000 ns | |
| Figure 5B (representative conformation of region B in Figure 4) | CMD_M2 | 1000 ns | |||
| Figure 5C (representative conformation of region A’ in Figure 4) | CMD_M3 | 1000 ns | |||
| Figure 5D (representative conformation of region B’ in Figure 4) | CMD_M4 | 1000 ns | |||
| Figure 5E (representative conformation of region C’ in Figure 4) | CMD_M5 | 1000 ns | |||
| Contact Number | Protein Tilt Angle | ΔGbinding | ΔGelec | ΔGvdw | ΔGpols | ΔGnpols | |
|---|---|---|---|---|---|---|---|
| CMD_M1 | 10,965 ± 671 | 29.0 ± 6.7 | −1894 ± 130 | −2420 ± 164 | −619 ± 44 | 1224 ± 98 | −79 ± 3 |
| CMD_M2 | 11,759 ± 658 | 16.7 ± 7.0 | −1933 ± 101 | −2506 ± 128 | −699 ± 38 | 1334 ± 158 | −61 ± 4 |
| CMD_M3 | 10,931 ± 660 | 30.9 ± 6.1 | −1812 ± 143 | −2045 ± 263 | −664 ± 42 | 979 ± 212 | −82 ± 4 |
| CMD_M4 | 11,547 ± 612 | 11.5 ± 4.5 | −1888 ± 115 | −2359 ± 134 | −661 ± 42 | 1193 ± 184 | −62 ± 6 |
| CMD_M5 | 10,317 ± 640 | 23.7 ± 7.0 | −1741 ± 130 | −1970 ± 163 | −624 ± 39 | 930 ± 178 | −78 ± 4 |
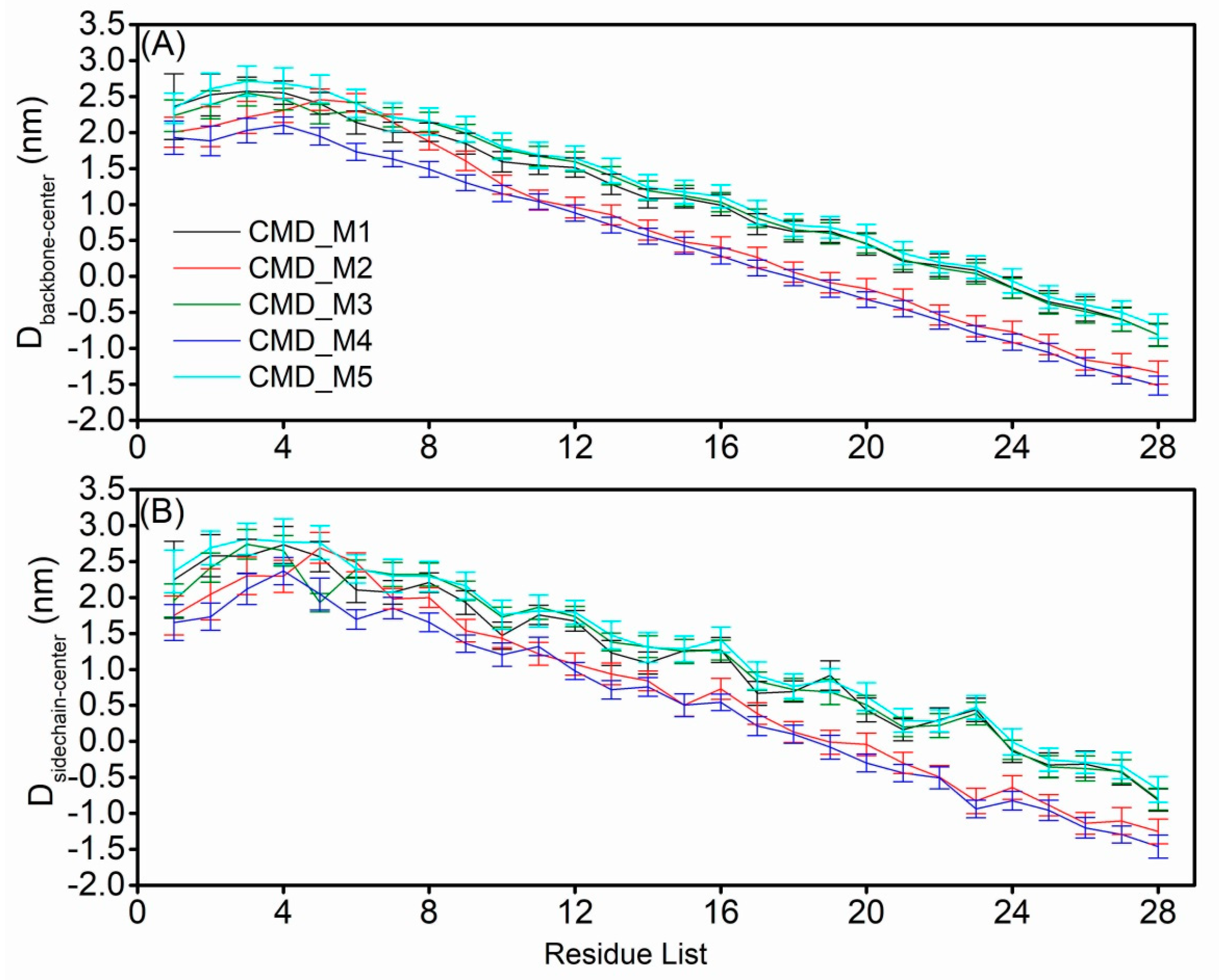
2.4. Structural Characteristics of the AapA1-28 Protein Based on CMD_M Simulations
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. MD Simulations for the AapA1-28 Tetramer-Containing Membrane Systems
5.2. D-MetaD Simulations for Single AapA1-28-Containing Membrane Systems
5.3. MD Simulations for Monomer AapA1-28-Containing Membrane Systems
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arnion, H.; Korkut, D.N.; Masachis Gelo, S.; Chabas, S.; Reignier, J.; Iost, I.; Darfeuille, F. Mechanistic insights into type I toxin antitoxin systems in Helicobacter pylori: The importance of mRNA folding in controlling toxin expression. Nucleic Acids Res. 2017, 45, 4782–4795. [Google Scholar]
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiss, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermuller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar] [CrossRef]
- Nonin-Lecomte, S.; Fermon, L.; Felden, B.; Pinel-Marie, M.L. Bacterial Type I Toxins: Folding and Membrane Interactions. Toxins 2021, 13, 490. [Google Scholar] [CrossRef] [PubMed]
- Korkut, D.N.; Alves, I.D.; Vogel, A.; Chabas, S.; Sharma, C.M.; Martinez, D.; Loquet, A.; Salgado, G.F.; Darfeuille, F. Structural insights into the AapA1 toxin of Helicobacter pylori. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129423. [Google Scholar] [CrossRef] [PubMed]
- El Mortaji, L.; Tejada-Arranz, A.; Rifflet, A.; Boneca, I.G.; Pehau-Arnaudet, G.; Radicella, J.P.; Marsin, S.; De Reuse, H. A peptide of a type I toxin-antitoxin system induces Helicobacter pylori morphological transformation from spiral shape to coccoids. Proc. Natl. Acad. Sci. USA 2020, 117, 31398–31409. [Google Scholar] [CrossRef]
- Sugita, M.; Sugiyama, S.; Fujie, T.; Yoshikawa, Y.; Yanagisawa, K.; Ohue, M.; Akiyama, Y. Large-Scale Membrane Permeability Prediction of Cyclic Peptides Crossing a Lipid Bilayer Based on Enhanced Sampling Molecular Dynamics Simulations. J. Chem. Inf. Model. 2021, 61, 3681–3695. [Google Scholar] [CrossRef]
- Kabelka, I.; Vacha, R. Advances in Molecular Understanding of alpha-Helical Membrane-Active Peptides. Acc. Chem. Res. 2021, 54, 2196–2204. [Google Scholar] [CrossRef]
- Ouyang, J.; Sheng, Y.; Wang, W. Recent Advances of Studies on Cell-Penetrating Peptides Based on Molecular Dynamics Simulations. Cells 2022, 11, 4016. [Google Scholar] [CrossRef]
- de Oliveira, E.C.L.; da Costa, K.S.; Taube, P.S.; Lima, A.H.; Junior, C.S.S. Biological Membrane-Penetrating Peptides: Computational Prediction and Applications. Front. Cell Infect. Microbiol. 2022, 12, 838259. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, T.; Prock, S.; Reichert, J.; Wadhwani, P.; Zimpfer, B.; Burck, J.; Berditsch, M.; Elstner, M.; Ulrich, A.S. Peptide-lipid interactions of the stress-response peptide TisB that induces bacterial persistence. Biophys. J. 2012, 103, 1460–1469. [Google Scholar] [CrossRef]
- Schneider, V.; Wadhwani, P.; Reichert, J.; Burck, J.; Elstner, M.; Ulrich, A.S.; Kubar, T. Tetrameric Charge-Zipper Assembly of the TisB Peptide in Membranes-Computer Simulation and Experiment. J. Phys. Chem. B 2019, 123, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Leveritt, J.M., 3rd; Pino-Angeles, A.; Lazaridis, T. The structure of a melittin-stabilized pore. Biophys. J. 2015, 108, 2424–2426. [Google Scholar] [CrossRef]
- Pino-Angeles, A.; Leveritt, J.M., 3rd; Lazaridis, T. Pore Structure and Synergy in Antimicrobial Peptides of the Magainin Family. PLoS Comput. Biol. 2016, 12, e1004570. [Google Scholar] [CrossRef] [PubMed]
- Chng, C.P.; Cho, N.J.; Hsia, K.J.; Huang, C. Role of Membrane Stretch in Adsorption of Antiviral Peptides onto Lipid Membranes and Membrane Pore Formation. Langmuir 2021, 37, 13390–13398. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, L.; Liu, L.; Yue, S.; Wang, J.; Cao, Z. Influence of Different Aromatic Hydrophobic Residues on the Antimicrobial Activity and Membrane Selectivity of BRBR-NH(2) Tetrapeptide. Langmuir 2020, 36, 15331–15342. [Google Scholar] [CrossRef]
- Yeasmin, R.; Buck, M.; Weinberg, A.; Zhang, L. Translocation of Human beta Defensin Type 3 through a Neutrally Charged Lipid Membrane: A Free Energy Study. J. Phys. Chem. B 2018, 122, 11883–11894. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, C.H.; Hu, D.; Ulmschneider, M.B.; Ulmschneider, J.P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016, 7, 13535. [Google Scholar] [CrossRef]
- Song, C.; de Groot, B.L.; Sansom, M.S.P. Lipid Bilayer Composition Influences the Activity of the Antimicrobial Peptide Dermcidin Channel. Biophys. J. 2019, 116, 1658–1666. [Google Scholar] [CrossRef]
- Kabelka, I.; Brozek, R.; Vacha, R. Selecting Collective Variables and Free-Energy Methods for Peptide Translocation across Membranes. J. Chem. Inf. Model. 2021, 61, 819–830. [Google Scholar] [CrossRef]
- Cao, Z.; Liu, L.; Hu, G.; Bian, Y.; Li, H.; Wang, J.; Zhou, Y. Interplay of hydrophobic and hydrophilic interactions in sequence-dependent cell penetration of spontaneous membrane-translocating peptides revealed by bias-exchange metadynamics simulations. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183402. [Google Scholar] [CrossRef]
- Hub, J.S. Joint Reaction Coordinate for Computing the Free-Energy Landscape of Pore Nucleation and Pore Expansion in Lipid Membranes. J. Chem. Theory Comput. 2021, 17, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.C.; Yusupov, M.; Cordeiro, R.M.; Bogaerts, A. Unraveling the permeation of reactive species across nitrated membranes by computer simulations. Comput. Biol. Med. 2021, 136, 104768. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Lv, H.; Guo, Y.; Peng, W.; Liu, B. sAMPpred-GAT: Prediction of antimicrobial peptide by graph attention network and predicted peptide structure. Bioinformatics 2023, 39, btac715. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.T.; Yang, L.Y.; Lu, I.H.; Cheng, W.C.; Hsu, Z.R.; Chen, S.H.; Lin, C.Y. AI4AMP: An Antimicrobial Peptide Predictor Using Physicochemical Property-Based Encoding Method and Deep Learning. Msystems 2021, 6, e0029921. [Google Scholar] [CrossRef]
- Lawrence, T.J.; Carper, D.L.; Spangler, M.K.; Carrell, A.A.; Rush, T.A.; Minter, S.J.; Weston, D.J.; Labbe, J.L. amPEPpy 1.0: A portable and accurate antimicrobial peptide prediction tool. Bioinformatics 2021, 37, 2058–2060. [Google Scholar] [CrossRef]
- Kawano, M.; Oshima, T.; Kasai, H.; Mori, H. Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol. Microbiol. 2002, 45, 333–349. [Google Scholar] [CrossRef]
- Pinel-Marie, M.L.; Brielle, R.; Felden, B. Dual toxic-peptide-coding Staphylococcus aureus RNA under antisense regulation targets host cells and bacterial rivals unequally. Cell Rep. 2014, 7, 424–435. [Google Scholar] [CrossRef]
- Jahn, N.; Preis, H.; Wiedemann, C.; Brantl, S. BsrG/SR4 from Bacillus subtilis—The first temperature-dependent type I toxin-antitoxin system. Mol. Microbiol. 2012, 83, 579–598. [Google Scholar] [CrossRef]
- Bogati, B.; Shore, S.F.H.; Nipper, T.D.; Stoiculescu, O.; Fozo, E.M. Charged Amino Acids Contribute to ZorO Toxicity. Toxins 2022, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Davila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- NosÉ, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 2002, 100, 191–198. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef]
- Kutzner, C.; Pall, S.; Fechner, M.; Esztermann, A.; de Groot, B.L.; Grubmuller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J Comput Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
| Prediction Methods | AapA1 (MATKHGKNSWKTLYLKISFLGCKVVVLLKR) | AapA1-28 (MATKHGKNSWKTLYLKISFLGCKVVVLL) |
|---|---|---|
| AMP_Scanner | AMP (0.98) | AMP (0.88) |
| sAMPpred-GAT | AMP (0.70) | nonAMP (0.09) |
| AI4AMP | AMP (0.60) | nonAMP (0.46) |
| amPEPpy 1.0 | nonAMP (0.29) | nonAMP (0.37) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; Zhao, L.; Yan, T.; Liu, L. Effects of C-Terminal Lys-Arg Residue of AapA1 Protein on Toxicity and Structural Mechanism. Toxins 2023, 15, 542. https://doi.org/10.3390/toxins15090542
Cao Z, Zhao L, Yan T, Liu L. Effects of C-Terminal Lys-Arg Residue of AapA1 Protein on Toxicity and Structural Mechanism. Toxins. 2023; 15(9):542. https://doi.org/10.3390/toxins15090542
Chicago/Turabian StyleCao, Zanxia, Liling Zhao, Tingting Yan, and Lei Liu. 2023. "Effects of C-Terminal Lys-Arg Residue of AapA1 Protein on Toxicity and Structural Mechanism" Toxins 15, no. 9: 542. https://doi.org/10.3390/toxins15090542