Continuous Treatment with IncobotulinumtoxinA Despite Presence of BoNT/A Neutralizing Antibodies: Immunological Hypothesis and a Case Report


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction and Background
1.1. Immunoresistance Is a Major Concern and Limitation of BoNT/A Therapy
1.2. Differences in Immunogenicity of BoNT/A Products Correlate with the Presence of Complexing Proteins and Other Bacterial Components
1.3. Breaking Immunoresistance Is Possible with a Low Immunogenic CPF-BoNT/A Product
1.4. Aims of Article
2. How the Immune System Is Activated—The Naïve Situation
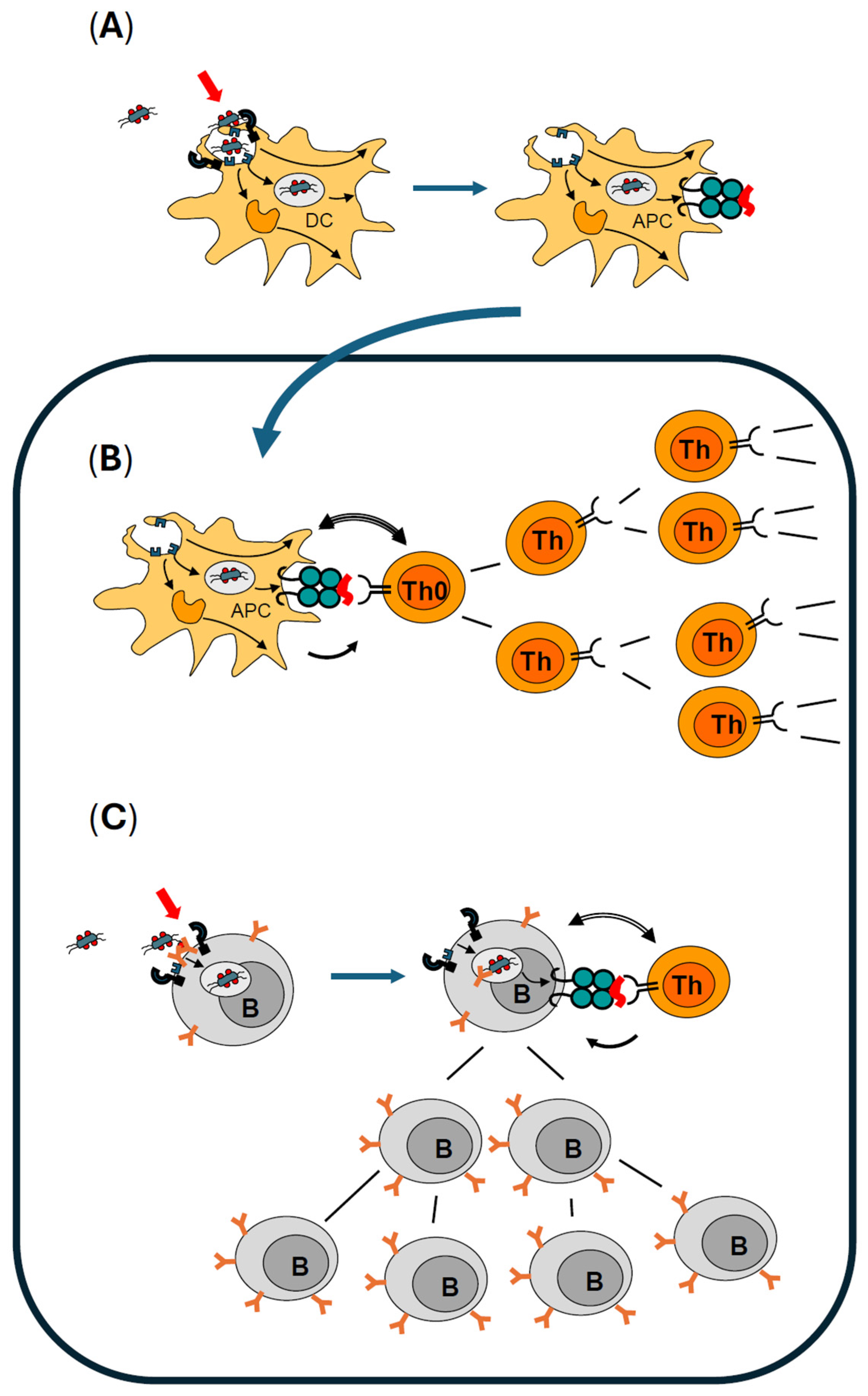
2.1. How the Immune System Becomes Activated to Produce Antibodies
2.2. Optimal Activation of Dendritic Cells Is Mandatory for Professional Antigen Presentation
2.3. Professional Antigen Presentation Is Mandatory for Activation and Clonal Expansion of Antigen-Specific Naïve T Helper Lymphocytes
2.4. Activation of Antigen-Specific B Lymphocytes Is Controlled by Antigen-Specific Effector T Helper Lymphocytes and Enhanced by Immunologic Adjuvants
2.5. B Effector Lymphocytes Have Several Options to Contribute to an Ongoing Immune Response
2.5.1. Immediate IgM Production and Release: Increasing the Quality of Defense Mechanisms by Specifically Directing Attack Mechanisms to the Labelled Surface of Microbes
2.5.2. Class Switching from IgM to IgG
2.5.3. Increasing Affinity While Maintaining Specificity: Somatic Hypermutation and Affinity Maturation
2.5.4. Antibodies Have a Short Half-Life and Need to Be Produced Continuously to Maintain an Effective Titer in the Blood
2.6. Response to BoNT/A Injections in the Naïve Situation
2.6.1. Activation of DCs by BoNT/A Injections
2.6.2. Presentation of BoNT/A-Derived Peptides to BoNT/A Peptide-Specific T Cells
2.6.3. What Is Known About B Cell Responses to BoNT/A?
2.6.4. Role of BoNT/A-Specific IgM or IgG Antibodies
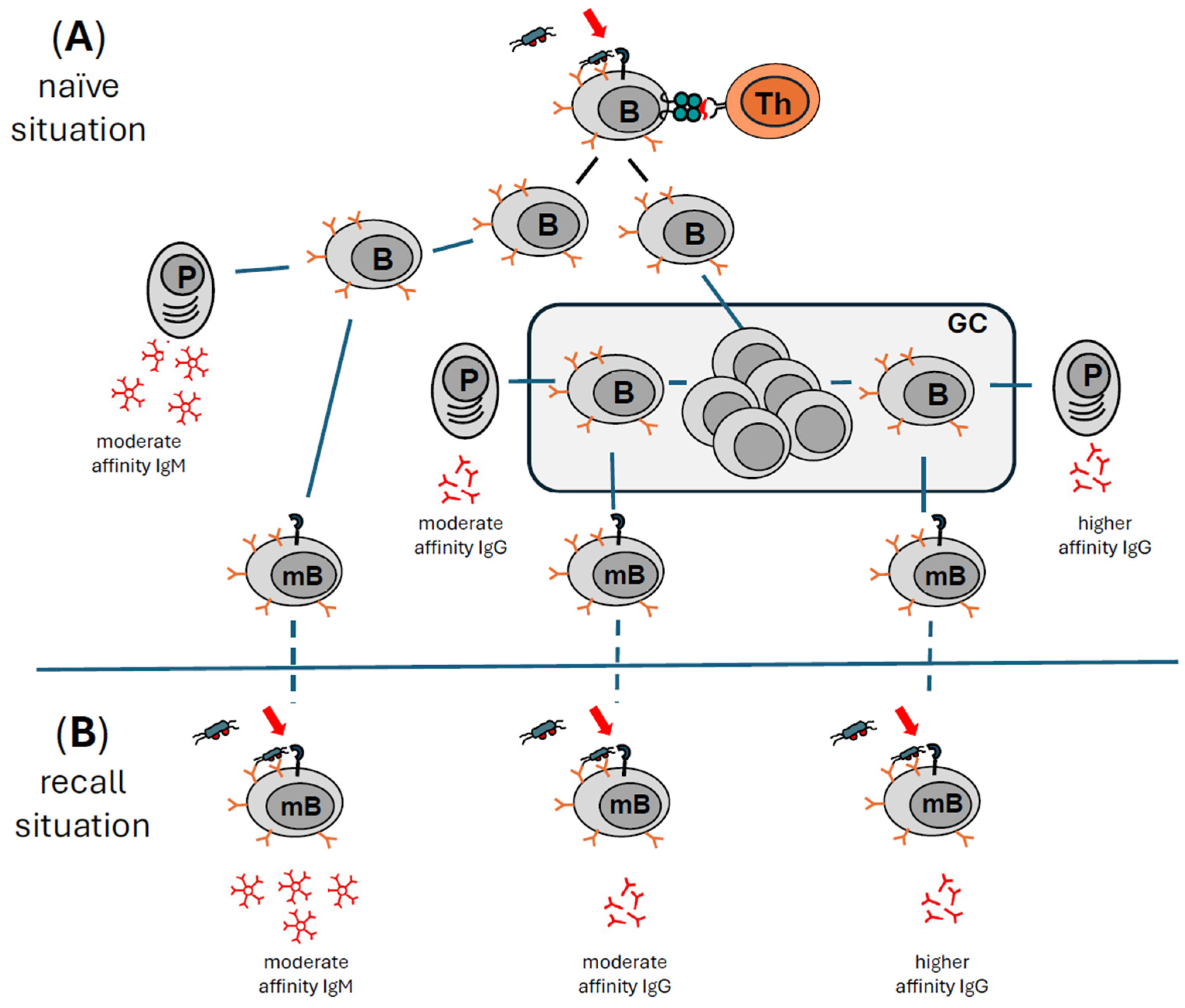
3. How the Immune System Remembers—The Recall Situation
3.1. The Innate Immune Arm Does Not Contribute to Long-Lived Memory
3.2. The Adaptive Arm of the Immune System Remembers
3.3. Different Types of B Cells Are Responsible for Prolonged Antibody Production
3.4. Reactivation of Memory B Cells Is More Effective in the Presence of Immunologic Adjuvants
3.5. Immunological Memory and Repeated Injections of BoNT/A—Role of Immunologic Adjuvants
4. Case Report
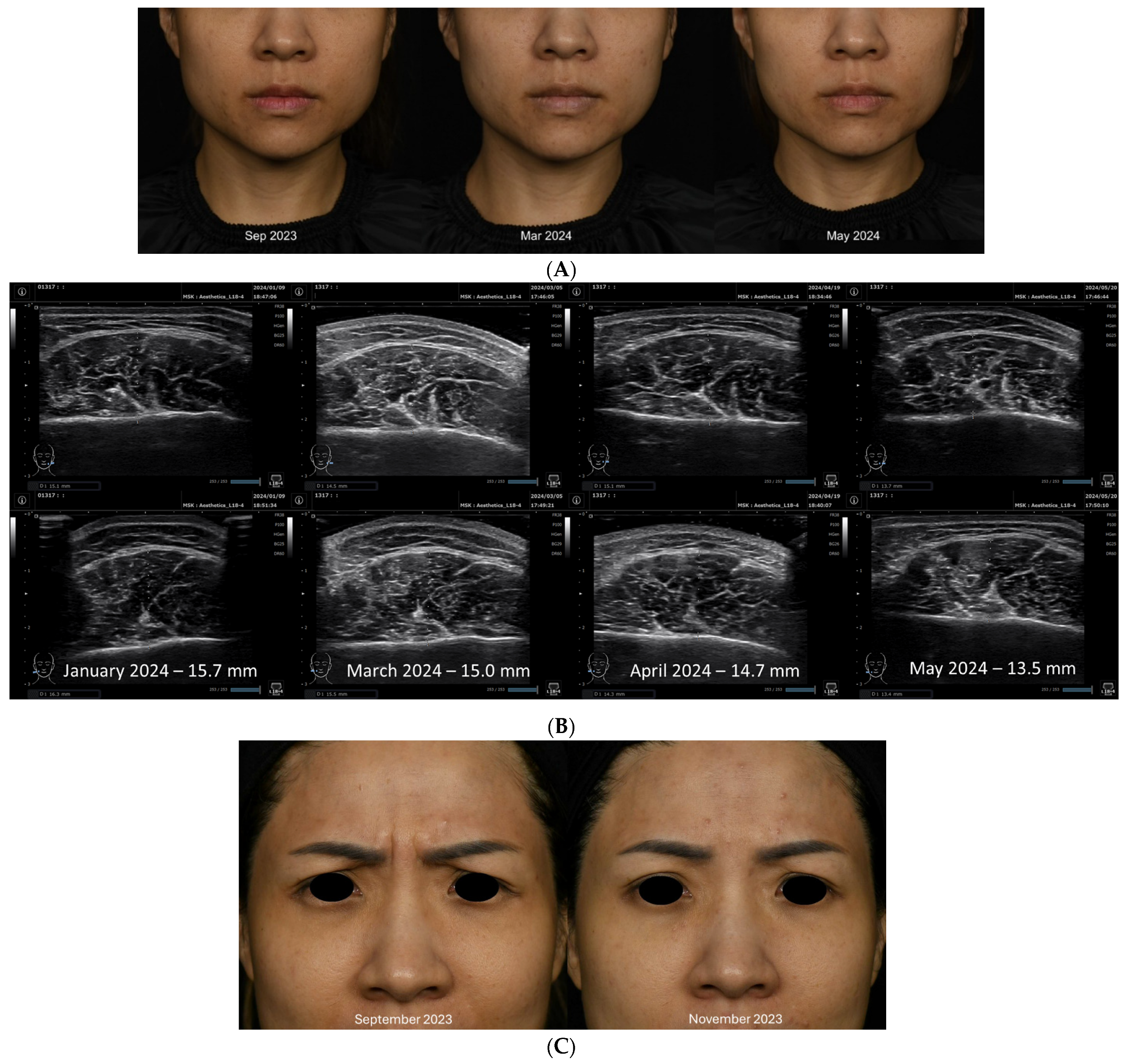
4.1. Clinical History
4.2. Summary of Case Report
5. Discussion
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jankovic, J.; Schwartz, K. Response and immunoresistance to botulinum toxin injections. Neurology 1995, 45, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D. Clinical presentation and management of antibody-induced failure of botulinum toxin therapy. Mov. Disord. 2004, 19 (Suppl. S8), S92–S100. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Leodori, G.; Fernandes, R.M.; Bhidayasiri, R.; Marti, M.J.; Colosimo, C.; Ferreira, J.J. Neutralizing Antibody and Botulinum Toxin Therapy: A Systematic Review and Meta-analysis. Neurotox. Res. 2016, 29, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Bellows, S.; Jankovic, J. Immunogenicity Associated with Botulinum Toxin Treatment. Toxins 2019, 11, 491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carr, W.W.; Jain, N.; Sublett, J.W. Immunogenicity of Botulinum Toxin Formulations: Potential Therapeutic Implications. Adv. Ther. 2021, 38, 5046–5064. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rahman, E.; Banerjee, P.S.; Asghar, A.; Gupta, N.K.; Mosahebi, A. Botulinum Toxin Type A Immunogenicity across Multiple Indications: An Overview Systematic Review. Plast. Reconstr. Surg. 2022, 149, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Carruthers, J.; Naumann, M.; Ogilvie, P.; Boodhoo, T.; Attar, M.; Gupta, S.; Singh, R.; Soliman, J.; Yushmanova, I.; et al. Neutralizing Antibody Formation with OnabotulinumtoxinA (BOTOX®) Treatment from Global Registration Studies across Multiple Indications: A Meta-Analysis. Toxins 2023, 15, 342. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dressler, D.; Wohlfahrt, K.; Meyer-Rogge, E.; Wiest, L.; Bigalke, H. Antibody-induced failure of botulinum toxin a therapy in cosmetic indications. Dermatol. Surg. 2010, 36 (Suppl. S4), 2182–2187. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Corduff, N.; Frevert, J.; Wanitphakdeedecha, R.; Chao, Y.Y.Y. Immunogenicity Associated with Aesthetic Botulinumtoxin A: A Survey of Asia-Pacific Physicians’ Experiences and Recommendations. Plast. Reconstr. Surg. Glob. Open 2022, 10, e4217. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ho, W.W.S.; Chan, L.; Corduff, N.; Lau, W.T.; Martin, M.U.; Tay, C.M.; Wang, S.; Wu, R. Addressing the Real-World Challenges of Immunoresistance to Botulinum Neurotoxin A in Aesthetic Practice: Insights and Recommendations from a Panel Discussion in Hong Kong. Toxins 2023, 15, 456. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Srinoulprasert, Y.; Wanitphakdeedecha, R. Antibody-induced botulinum toxin treatment failure: A review and novel management approach. J. Cosmet. Dermatol. 2020, 19, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Marion, M.H.; Humberstone, M.; Grunewald, R.; Wimalaratna, S. British Neurotoxin Network recommendations for managing cervical dystonia in patients with a poor response to botulinum toxin. Pract. Neurol. 2016, 16, 288–295. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dressler, D.; Bigalke, H.; Benecke, R. Botulinum toxin type B in antibody-induced botulinum toxin type A therapy failure. J. Neurol. 2003, 250, 967–969. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D.; Bigalke, H. Botulinum toxin antibody type A titres after cessation of botulinum toxin therapy. Mov. Disord. 2002, 17, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Pirazzini, M.; Montecucco, C. Botulinum neurotoxins: Genetic, structural and mechanistic insights. Nat. Rev. Microbiol. 2014, 12, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frevert, J. Content of botulinum neurotoxin in Botox®/Vistabel®, Dysport®/Azzalure®, and Xeomin®/Bocouture®. Drugs R D 2010, 10, 67–73. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frevert, J. Pharmaceutical, biological, and clinical properties of botulinum neurotoxin type A products. Drugs R D 2015, 15, 1–9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- FDA Center for Evaluation and Research 2009. “Dysport/AbobotulinumtoxinA”. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/125274Orig1s000MedR.pdf (accessed on 18 June 2024).
- FDA Prescribing Information, Botox/OnabotulinumtoxinA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/103000s5236lbl.pdf (accessed on 18 June 2024).
- FDAPrescribing Information, Xeomin/IncobotulinumtoxinA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125360s073lbl.pdf (accessed on 7 August 2024).
- Dressler, D. Five-year experience with incobotulinumtoxinA (Xeomin(®)): The first botulinum toxin drug free of complexing proteins. Eur. J. Neurol. 2012, 19, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Frevert, J. Xeomin is free from complexing proteins. Toxicon 2009, 54, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, M.; Wanitphakdeedecha, R.; Trindade de Almeida, A.; Maas, C.; Frevert, J. IncobotulinumtoxinA: A Highly Purified and Precisely Manufactured Botulinum Neurotoxin Type A. J. Drugs Dermatol. 2019, 18, 52–57. [Google Scholar] [PubMed]
- Panjwani, N.; O‘Keeffe, R.; Pickett, A. Biochemical, functional and potency characteristics of type A botulinum toxin in clinical use. Botulinum J. 2008, 1, 153–166. [Google Scholar] [CrossRef]
- Frevert, J.; Grönewald, C. Presence of clostridial DNA in botulinum toxin products. Toxicon 2015, 93, S28. [Google Scholar] [CrossRef]
- Hefter, H.; Rosenthal, D.; Jansen, A.; Brauns, R.; Ürer, B.; Bigalke, H.; Hartung, H.P.; Meuth, S.G.; Lee, J.I.; Albrecht, P.; et al. Significantly lower antigenicity of incobotulinumtoxin than abo- or onabotulinumtoxin. J. Neurol. 2023, 270, 788–796. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wissel, J.; Bensmail, D.; Ferreira, J.J.; Molteni, F.; Satkunam, L.; Moraleda, S.; Rekand, T.; McGuire, J.; Scheschonka, A.; Flatau-Baqué, B.; et al. Safety and efficacy of incobotulinumtoxinA doses up to 800 U in limb spasticity: The TOWER study. Neurology 2017, 88, 1321–1328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Albrecht, P.; Jansen, A.; Lee, J.I.; Moll, M.; Ringelstein, M.; Rosenthal, D.; Bigalke, H.; Aktas, O.; Hartung, H.P.; Hefter, H. High prevalence of neutralizing antibodies after long-term botulinum neurotoxin therapy. Neurology 2019, 92, e48–e54, Erratum in: Neurology 2022, 98, 341. [Google Scholar] [CrossRef] [PubMed]
- Walter, U.; Mühlenhoff, C.; Benecke, R.; Dressler, D.; Mix, E.; Alt, J.; Wittstock, M.; Dudesek, A.; Storch, A.; Kamm, C. Frequency and risk factors of antibody-induced secondary failure of botulinum neurotoxin therapy. Neurology 2020, 94, e2109–e2120, Erratum in: Neurology 2020, 95, 802. [Google Scholar] [CrossRef] [PubMed]
- Hefter, H.; Brauns, R.; Ürer, B.; Rosenthal, D.; Albrecht, P. Effective long-term treatment with incobotulinumtoxin (Xeomin®) without neutralizing antibody induction: A monocentric, cross-sectional study. J. Neurol. 2020, 267, 1340–1347. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Samadzadeh, S.; Ürer, B.; Brauns, R.; Rosenthal, D.; Lee, J.I.; Albrecht, P.; Hefter, H. Clinical Implications of Difference in Antigenicity of Different Botulinum Neurotoxin Type A Preparations: Clinical Take-Home Messages from Our Research Pool and Literature. Toxins 2020, 12, 499. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hefter, H.; Ürer, B.; Brauns, R.; Rosenthal, D.; Meuth, S.G.; Lee, J.I.; Albrecht, P.; Samadzadeh, S. Significant Long-Lasting Improvement after Switch to Incobotulinum Toxin in Cervical Dystonia Patients with Secondary Treatment Failure. Toxins 2022, 14, 44. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dressler, D. Antikörper vermitteltes Versagen der Botulinum-Toxin-Therapie [Antibody-induced failure of botulinum toxin therapy]. Nervenarzt 2003, 74, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D.; Pan, L.; Adib Saberi, F. Antibody-induced failure of botulinum toxin therapy: Re-start with low-antigenicity drugs offers a new treatment opportunity. J. Neural Transm. 2018, 125, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Hefter, H.; Hartmann, C.; Kahlen, U.; Moll, M.; Bigalke, H. Prospective analysis of neutralising antibody titres in secondary non-responders under continuous treatment with a botulinumtoxin type A preparation free of complexing proteins—A single cohort 4-year follow-up study. BMJ Open 2012, 2, e000646. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bulbringe, E. Observations on the isolated phrenic nerve diaphragm preparation of the rat. Br. J. Pharmacol. Chemother. 1946, 1, 38–61. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rasetti-Escargueil, C.; Liu, Y.; Rigsby, P.; Jones, R.G.; Sesardic, D. Phrenic nerve-hemidiaphragm as a highly sensitive replacement assay for determination of functional botulinum toxin antibodies. Toxicon 2011, 57, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, H.; Rummel, A. Botulinum Neurotoxins: Qualitative and Quantitative Analysis Using the Mouse Phrenic Nerve Hemidiaphragm Assay (MPN). Toxins 2015, 7, 4895–4905. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dressler, D.; Adib Saberi, F.; Bigalke, H. IncobotulinumtoxinA (Xeomin(®)) can produce antibody-induced therapy failure in a patient pretreated with abobotulinumtoxinA (Dysport(®)). J. Neural Transm. 2014, 121, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Hefter, H.; Hartmann, C.J.; Kahlen, U.; Samadzadeh, S.; Rosenthal, D.; Moll, M. Clinical Improvement After Treatment With IncobotulinumtoxinA (XEOMIN®) in Patients With Cervical Dystonia Resistant to Botulinum Toxin Preparations Containing Complexing Proteins. Front. Neurol. 2021, 12, 636590. [Google Scholar] [CrossRef] [PubMed]
- Ramos, V.F.; Karp, B.I.; Lungu, C.; Alter, K.; Hallett, M. Clinical Response to IncobotulinumtoxinA, after Demonstrated Loss of Clinical Response to OnabotulinumtoxinA and RimabotulininumtoxinB in a Patient with Musician’s Dystonia. Mov. Disord. Clin. Pract. 2014, 1, 383–385. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Hilligan, K.L.; Ronchese, F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Pattern recognition receptors: Doubling up for the innate immune response. Cell 2002, 111, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C., Jr. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Pradeu, T.; Cooper, E.L. The danger theory: 20 years later. Front. Immunol. 2012, 3, 287. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wikipedia Definition of “Immunologic Adjuvant”. Available online: https://en.wikipedia.org/wiki/Immunologic_adjuvant (accessed on 18 June 2024).
- Dzopalic, T.; Rajkovic, I.; Dragicevic, A.; Colic, M. The response of human dendritic cells to co-ligation of pattern-recognition receptors. Immunol. Res. 2012, 52, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Zhang, X.; Cheng, Y.; Cao, X. Dendritic cell migration in inflammation and immunity. Cell Mol. Immunol. 2021, 18, 2461–2471. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sprent, J.; Surh, C.D. Generation and maintenance of memory T cells. Curr. Opin. Immunol. 2001, 13, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Marchingo, J.M.; Horton, M.B.; Hodgkin, P.D. The regulation of lymphocyte activation and proliferation. Curr. Opin. Immunol. 2018, 51, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Rickert, R.C. Regulation of B lymphocyte activation by complement C3 and the B cell coreceptor complex. Curr. Opin. Immunol. 2005, 17, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.J.; Banerjee, A. Regulating B-cell activation and survival in response to TLR signals. Immunol. Cell Biol. 2007, 85, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Hou, B. TLR signaling in B-cell development and activation. Cell Mol. Immunol. 2013, 10, 103–106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Z.; Wang, J.H. How the Signaling Crosstalk of B Cell Receptor (BCR) and Co-Receptors Regulates Antibody Class Switch Recombination: A New Perspective of Checkpoints of BCR Signaling. Front. Immunol. 2021, 12, 663443. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Y.; Garcia-Ibanez, L.; Toellner, K.M. Regulation of germinal center B-cell differentiation. Immunol. Rev. 2016, 270, 8–19, Erratum in: Immunol. Rev. 2016, 272, 202. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 2005, 23, 487–513. [Google Scholar] [CrossRef] [PubMed]
- Budeus, B.; Kibler, A.; Küppers, R. Human IgM-expressing memory B cells. Front. Immunol. 2023, 14, 1308378. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Victora, G.D.; Nussenzweig, M.C. Germinal Centers. Annu. Rev. Immunol. 2022, 40, 413–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zan, H.; Pone, E.J.; Mai, T.; Casali, P. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat. Rev. Immunol. 2012, 12, 517–531. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brynjolfsson, S.F.; Persson Berg, L.; Olsen Ekerhult, T.; Rimkute, I.; Wick, M.J.; Mårtensson, I.L.; Grimsholm, O. Long-Lived Plasma Cells in Mice and Men. Front. Immunol. 2018, 9, 2673. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lightman, S.M.; Utley, A.; Lee, K.P. Survival of Long-Lived Plasma Cells (LLPC): Piecing Together the Puzzle. Front. Immunol. 2019, 10, 965. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Palm, A.E.; Henry, C. Remembrance of Things Past: Long-Term B Cell Memory After Infection and Vaccination. Front. Immunol. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jego, G.; Pascual, V.; Palucka, A.K.; Banchereau, J. Dendritic cells control B cell growth and differentiation. Curr. Dir. Autoimmun. 2005, 8, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A.; Bernasconi, N.; Traggiai, E.; Ruprecht, C.R.; Corti, D.; Sallusto, F. Understanding and making use of human memory B cells. Immunol. Rev. 2006, 211, 303–309. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ueno, H.; Schmitt, N.; Palucka, A.K.; Banchereau, J. Dendritic cells and humoral immunity in humans. Immunol. Cell Biol. 2010, 88, 376–380. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, W.; Banchereau, J. The antigen presenting cells instruct plasma cell differentiation. Front. Immunol. 2014, 4, 504. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jain, A.; Pasare, C. Innate Control of Adaptive Immunity: Beyond the Three-Signal Paradigm. J. Immunol. 2017, 198, 3791–3800. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carroll, S.L.; Pasare, C.; Barton, G.M. Control of adaptive immunity by pattern recognition receptors. Immunity 2024, 57, 632–648. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amanna, I.J.; Slifka, M.K. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol. Rev. 2010, 236, 125–138, Erratum in: Immunol. Rev. 2010, 237, 284. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mizel, S.B.; Bates, J.T. Flagellin as an adjuvant: Cellular mechanisms and potential. J. Immunol. 2010, 185, 5677–5682. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cui, B.; Liu, X.; Fang, Y.; Zhou, P.; Zhang, Y.; Wang, Y. Flagellin as a vaccine adjuvant. Expert Rev. Vaccines. 2018, 17, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Paludan, S.R.; Fitzgerald, K.A. DNA recognition in immunity and disease. Curr. Opin. Immunol. 2013, 25, 13–18. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bauer, S.; Wagner, H. Bacterial CpG-DNA licenses TLR9. Curr. Top Microbiol. Immunol. 2002, 270, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Kayraklioglu, N.; Horuluoglu, B.; Klinman, D.M. CpG Oligonucleotides as Vaccine Adjuvants. Methods Mol. Biol. 2021, 2197, 51–85. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Singh, B.R. Immunological properties of Hn-33 purified from type A Clostridium botulinum. J. Nat. Toxins. 2000, 9, 357–362. [Google Scholar] [PubMed]
- Mahmut, N.; Inoue, K.; Fujinaga, Y.; Hughes, L.; Arimitsu, H.; Sakaguchi, Y.; Ohtsuka, A.; Murakami, T.; Yokota, K.; Oguma, K. Characterisation of monoclonal antibodies against haemagglutinin associated with Clostridium botulinum type C neurotoxin. J. Med. Microbiol. 2002, 51, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Yokota, K.; Arimitsu, H.; Hwang, H.J.; Sakaguchi, Y.; Cui, J.; Takeshi, K.; Watanabe, T.; Ohyama, T.; Oguma, K. Production of anti-neurotoxin antibody is enhanced by two subcomponents, HA1 and HA3b, of Clostridium botulinum type B 16S toxin-haemagglutinin. Microbiology 2005, 151 Pt 11, 3739–3747. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R.; Chang, T.W.; Cai, S.; Lindo, P.; Riding, S.; Zhou, Y.; Ravichandran, E.; Singh, B.R. Immunological characterization of the subunits of type A botulinum neurotoxin and different components of its associated proteins. Toxicon 2009, 53, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Sayadmanesh, A.; Ebrahimi, F.; Hajizade, A.; Rostamian, M.; Keshavarz, H. Expression and purification of neurotoxin-associated protein HA-33/A from Clostridium botulinum and evaluation of its antigenicity. Iran. Biomed. J. 2013, 17, 165–170. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bryant, A.M.; Cai, S.; Singh, B.R. Comparative immunochemical characteristics of botulinum neurotoxin type A and its associated proteins. Toxicon 2013, 72, 126–132. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, L.; Sun, Y.; Yang, W.; Lindo, P.; Singh, B.R. Type A botulinum neurotoxin complex proteins differentially modulate host response of neuronal cells. Toxicon 2014, 82, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S. Effects of protein aggregates: An immunologic perspective. AAPS J. 2006, 8, E501–E507. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- den Engelsman, J.; Garidel, P.; Smulders, R.; Koll, H.; Smith, B.; Bassarab, S.; Seidl, A.; Hainzl, O.; Jiskoot, W. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm. Res. 2011, 28, 920–933. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pham, N.B.; Meng, W.S. Protein aggregation and immunogenicity of biotherapeutics. Int. J. Pharm. 2020, 585, 119523. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Manfredi, A.A.; Capobianco, A.; Bianchi, M.E.; Rovere-Querini, P. Regulation of dendritic- and T-cell fate by injury-associated endogenous signals. Crit. Rev. Immunol. 2009, 29, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Nace, G.; Evankovich, J.; Eid, R.; Tsung, A. Dendritic cells and damage-associated molecular patterns: Endogenous danger signals linking innate and adaptive immunity. J. Innate Immun. 2012, 4, 6–15. [Google Scholar] [CrossRef] [PubMed]
- White, D.M.; Pellett, S.; Jensen, M.A.; Tepp, W.H.; Johnson, E.A.; Arnason, B.G. Rapid immune responses to a botulinum neurotoxin Hc subunit vaccine through in vivo targeting to antigen-presenting cells. Infect. Immun. 2011, 79, 3388–3396. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Atassi, M.Z. Basic immunological aspects of botulinum toxin therapy. Mov. Disord. 2004, 19 (Suppl. S8), S68–S84. [Google Scholar] [CrossRef] [PubMed]
- Atassi, M.Z. Molecular basis of immunogenicity to botulinum neurotoxins and uses of the defined antigenic regions. Toxicon 2015, 107 Pt A, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Hayakari, M.; Middlebrook, J.L.; Atassi, M.Z. Immune recognition of botulinum neurotoxin type A: Regions recognized by T cells and antibodies against the protective H(C) fragment (residues 855-1296) of the toxin. Mol. Immunol. 1997, 34, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Dolimbek, B.Z.; Steward, L.E.; Aoki, K.R.; Atassi, M.Z. Regions of recognition by blocking antibodies on the light chain of botulinum neurotoxin A: Antigenic structure of the entire toxin. Immunobiology 2011, 216, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Deitiker, P.; Jankovic, J.; Atassi, M.Z. Submolecular recognition regions of the HN domain of the heavy chain of botulinum neurotoxin type A by T cells from toxin-treated cervical dystonia patients. J. Neuroimmunol. 2016, 300, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Deitiker, P.; Jankovic, J.; Atassi, M.Z. The Regions on the Light Chain of Botulinum Neurotoxin Type A Recognized by T Cells from Toxin-Treated Cervical Dystonia Patients. The Complete Human T-Cell Recognition Map of the Toxin Molecule. Immunol. Investig. 2018, 47, 18–39. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Deitiker, P.R.; Jankovic, J.; Duane, D.D.; Aoki, K.R.; Atassi, M.Z. Human T-cell responses to botulinum neurotoxin: Proliferative responses in vitro of lymphocytes from botulinum neurotoxin A-treated movement disorder patients. J. Neuroimmunol. 2011, 237, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Deitiker, P.; Oshima, M.; Jankovic, J.; Atassi, M.Z. Influences of HLA DRB1, DQA1 and DQB1 on T-cell recognition of epitopes and of larger regions of the botulinum neurotoxin molecule. Immunol. Lett. 2017, 190, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Vuong, K.D.; Ahsan, J. Comparison of efficacy and immunogenicity of original versus current botulinum toxin in cervical dystonia. Neurology. 2003, 60, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D.; Saberi, F.A. New formulation of Botox: Complete antibody-induced treatment failure in cervical dystonia. BMJ Case Rep. 2009, 2009, bcr08.2008.0611. [Google Scholar] [CrossRef] [PubMed]
- Benecke, R. Clinical relevance of botulinum toxin immunogenicity. BioDrugs 2012, 26, e1–e9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galazka, A.; Rymkiewicz, D.; Aleksandrowicz, J. Botulinum antitoxins and antibacterial IgM and IgG antibodies in sera of persons immunized with botulinum polytoxoid combined with cholera vaccine. I. Response to botulinum toxoid. Arch. Immunol. Ther. Exp. 1976, 24, 631–639. [Google Scholar] [PubMed]
- Galazka, A.; Rymkiewicz, D.; Aleksandrowicz, J. Botulinum antitoxins and antibacterial IgM and IgG antibodies in sera of persons immunized with botulinum polytoxoid combined with cholera vaccine. II. Response to cholera vaccine. Arch. Immunol. Ther. Exp. 1976, 24, 641–654. [Google Scholar] [PubMed]
- Galazka, A.; Rymkiewicz, D.; Aleksandrowicz, J. Immunological response to Clostridium botulinum toxin. J. Clin. Microbiol. 1988, 26, 1250–1251. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Adekar, S.P.; Al-Saleem, F.H.; Elias, M.D.; Rybinski, K.A.; Simpson, L.L.; Dessain, S.K. A natural human IgM antibody that neutralizes botulinum neurotoxin in vivo. Hybridoma 2008, 27, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.E. Characterization of new formalin-detoxified botulinum neurotoxin toxoids. Clin. Vaccine Immunol. 2008, 15, 1374–1379. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral immunity due to long-lived plasma cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; SArunachalam, P.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pasare, C.; Medzhitov, R. Control of B-cell responses by Toll-like receptors. Nature 2005, 438, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Kasturi, S.P.; Skountzou, I.; Albrecht, R.A.; Koutsonanos, D.; Hua, T.; Nakaya, H.I.; Ravindran, R.; Stewart, S.; Alam, M.; Kwissa, M.; et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 2011, 470, 543–547. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meyer-Bahlburg, A.; Khim, S.; Rawlings, D.J. B cell intrinsic TLR signals amplify but are not required for humoral immunity. J. Exp. Med. 2007, 204, 3095–3101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richard, K.; Pierce, S.K.; Song, W. The agonists of TLR4 and 9 are sufficient to activate memory B cells to differentiate into plasma cells in vitro but not in vivo. J. Immunol. 2008, 181, 1746–1752. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rawlings, D.J.; Schwartz, M.A.; Jackson, S.W.; Meyer-Bahlburg, A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat. Rev. Immunol. 2012, 12, 282–294. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meyer-Bahlburg, A.; Rawlings, D.J. Differential impact of Toll-like receptor signaling on distinct B cell subpopulations. Front. Biosci. (Landmark Ed.) 2012, 17, 1499–1516. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Hosono, A.; Odagiri, T.; Tashiro, M.; Kaminogawa, S.; Okuno, Y.; Kurosaki, T.; Ato, M.; Kobayashi, K.; Takahashi, Y. Whole-Virion Influenza Vaccine Recalls an Early Burst of High-Affinity Memory B Cell Response through TLR Signaling. J. Immunol. 2016, 196, 4172–4184. [Google Scholar] [CrossRef] [PubMed]
- Brin, M.F.; James, C.; Maltman, J. Botulinum toxin type A products are not interchangeable: A review of the evidence. Biologics. 2014, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Samizadeh, S.; De Boulle, K. Botulinum neurotoxin formulations: Overcoming the confusion. Clin. Cosmet. Investig. Dermatol. 2018, 11, 273–287, Erratum in: Clin. Cosmet. Investig. Dermatol. 2018, 11, 629. [Google Scholar] [CrossRef] [PubMed]
- Brin, M.F.; Nelson, M.; Ashourian, N.; Brideau-Andersen, A.; Maltman, J. Update on Non-Interchangeability of Botulinum Neurotoxin Products. Toxins 2024, 16, 266. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- FDA Prescribing Information DAXXIFY/DaxibotulinumtoxinA-Lanm. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761127s000lbl.pdf (accessed on 7 August 2024).
- Medytox Homepage. Available online: https://www.medytox.com/page/coretox_en?site_id=en (accessed on 6 December 2023).
- TGAeBS-Product and Consumer Information. Available online: https://www.ebs.tga.gov.au/ebs/picmi/picmirepository.nsf/pdf?OpenAgent=&id=CP-2024-PI-02196-1&d=20240807172310101 (accessed on 26 September 2024).
- Press Release Galderma on, RelabotulinumtoxinA. Available online: https://www.galderma.com/news/galdermas-relfydesstm-relabotulinumtoxina-receives-positive-decision-use-europe (accessed on 7 August 2024).
- Sanz, J.; Randolph, H.E.; Barreiro, L.B. Genetic and evolutionary determinants of human population variation in immune responses. Curr. Opin. Genet. Dev. 2018, 53, 28–35. [Google Scholar] [CrossRef] [PubMed]
- “The Major Histocompatibility Complex and Its Function” in Immunobiology: The Immune System in Health and Disease. Available online: https://www.ncbi.nlm.nih.gov/books/NBK27156/ (accessed on 18 June 2024).
- Wenger, M.; Grosse-Kathoefer, S.; Kraiem, A.; Pelamatti, E.; Nunes, N.; Pointner, L.; Aglas, L. When the allergy alarm bells toll: The role of Toll-like receptors in allergic diseases and treatment. Front. Mol. Biosci. 2023, 10, 1204025. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nepal, M.R.; Jeong, T.C. Alternative Methods for Testing Botulinum Toxin: Current Status and Future Perspectives. Biomol. Ther. 2020, 28, 302–310. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dressler, D.; Rothwell, J.C.; Bhatia, K.; Kopp, B.; Bigalke, H.; Adib Saberi, F. Botulinum toxin antibody titres: Measurement, interpretation, and practical recommendations. J. Neurol. 2023, 270, 1524–1530. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sesardic, D.; Jones, R.G.; Leung, T.; Alsop, T.; Tierney, R. Detection of antibodies against botulinum toxins. Mov. Disord. 2004, 19 (Suppl. S8), S85–S91. [Google Scholar] [CrossRef] [PubMed]
- Hefter, H.; Rosenthal, D.; Moll, M. High Botulinum Toxin-Neutralizing Antibody Prevalence Under Long-Term Cervical Dystonia Treatment. Mov. Disord. Clin. Pract. 2016, 3, 500–506. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dressler, D.; Gessler, F.; Tacik, P.; Bigalke, H. An enzyme-linked immunosorbent assay for detection of botulinum toxin-antibodies. Mov. Disord. 2014, 29, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Srinoulprasert, Y.; Kantaviro, W.; Nokdhes, Y.N.; Patthamalai, P.; Dowdon, L.; Chawengkiattikul, R.; Wanitphakdeedecha, R. Development of inhibition ELISA to detect antibody-induced failure of botulinum toxin a therapy in cosmetic indications. J. Immunol. Methods 2019, 473, 112635. [Google Scholar] [CrossRef] [PubMed]
- Kranz, G.; Sycha, T.; Voller, B.; Kranz, G.S.; Schnider, P.; Auff, E. Neutralizing antibodies in dystonic patients who still respond well to botulinum toxin type A. Neurology 2008, 70, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Lange, O.; Bigalke, H.; Dengler, R.; Wegner, F.; deGroot, M.; Wohlfarth, K. Neutralizing antibodies and secondary therapy failure after treatment with botulinum toxin type A: Much ado about nothing? Clin. Neuropharmacol. 2009, 32, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Boo, L.M.; Ackerman, A.H.; Gallagher, C.J. Immunogenicity of botulinum toxins. J. Neural Transm. 2013, 120, 275–290. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, M.U.; Tay, C.M.; Siew, T.W. Continuous Treatment with IncobotulinumtoxinA Despite Presence of BoNT/A Neutralizing Antibodies: Immunological Hypothesis and a Case Report. Toxins 2024, 16, 422. https://doi.org/10.3390/toxins16100422
Martin MU, Tay CM, Siew TW. Continuous Treatment with IncobotulinumtoxinA Despite Presence of BoNT/A Neutralizing Antibodies: Immunological Hypothesis and a Case Report. Toxins. 2024; 16(10):422. https://doi.org/10.3390/toxins16100422
Chicago/Turabian StyleMartin, Michael Uwe, Clifton Ming Tay, and Tuck Wah Siew. 2024. "Continuous Treatment with IncobotulinumtoxinA Despite Presence of BoNT/A Neutralizing Antibodies: Immunological Hypothesis and a Case Report" Toxins 16, no. 10: 422. https://doi.org/10.3390/toxins16100422
APA StyleMartin, M. U., Tay, C. M., & Siew, T. W. (2024). Continuous Treatment with IncobotulinumtoxinA Despite Presence of BoNT/A Neutralizing Antibodies: Immunological Hypothesis and a Case Report. Toxins, 16(10), 422. https://doi.org/10.3390/toxins16100422