In Silico Analysis of Shiga Toxin-Producing Escherichia coli O157:H7 Strains from Presumptive Super- and Low-Shedder Cattle

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Results
2.1. In Silico Detection of Serotype, Multi-Locus Sequence Typing (MLST), Virulence Genes, Insertion Sites, Lineage, and Clade Profile in STEC O157
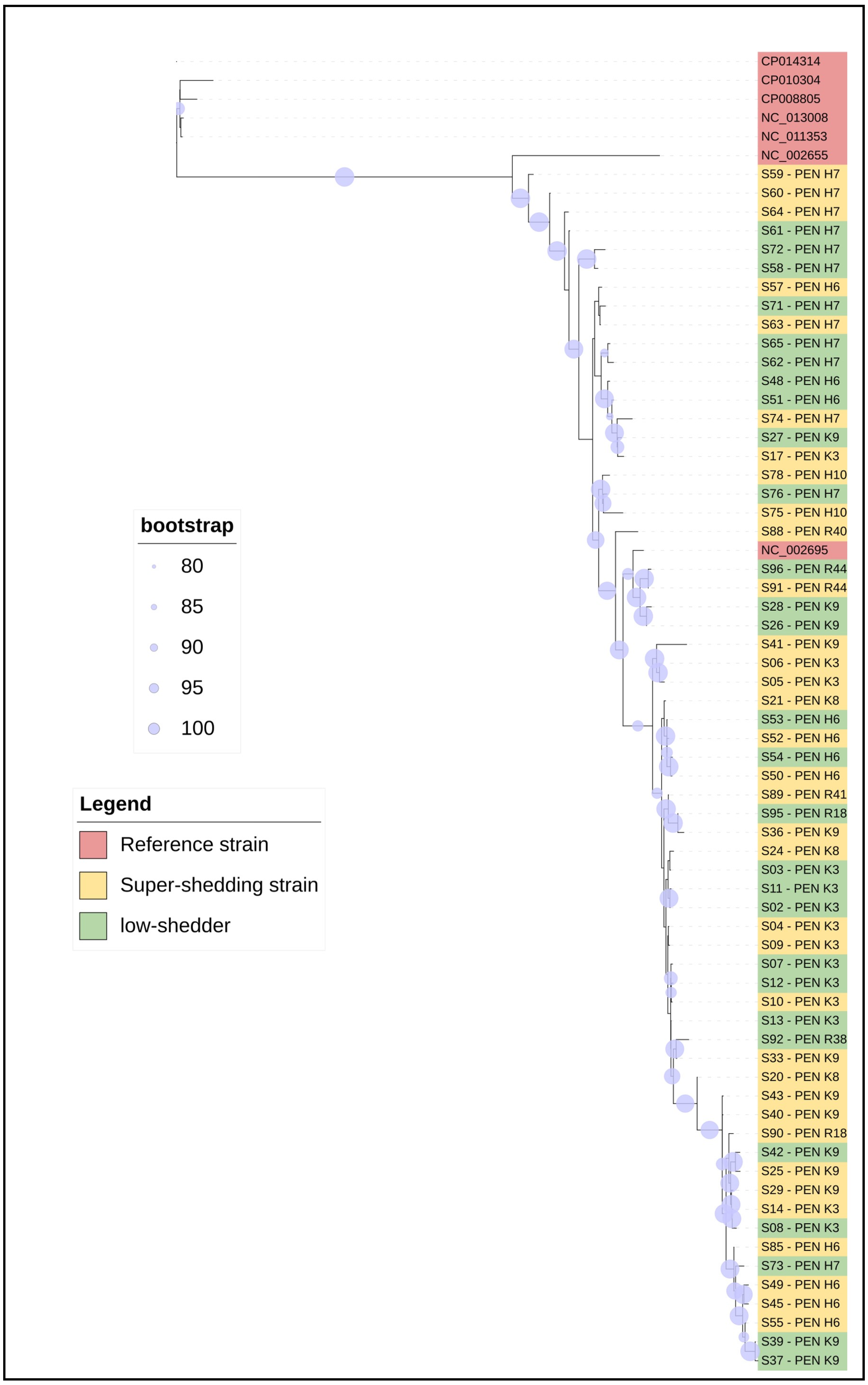
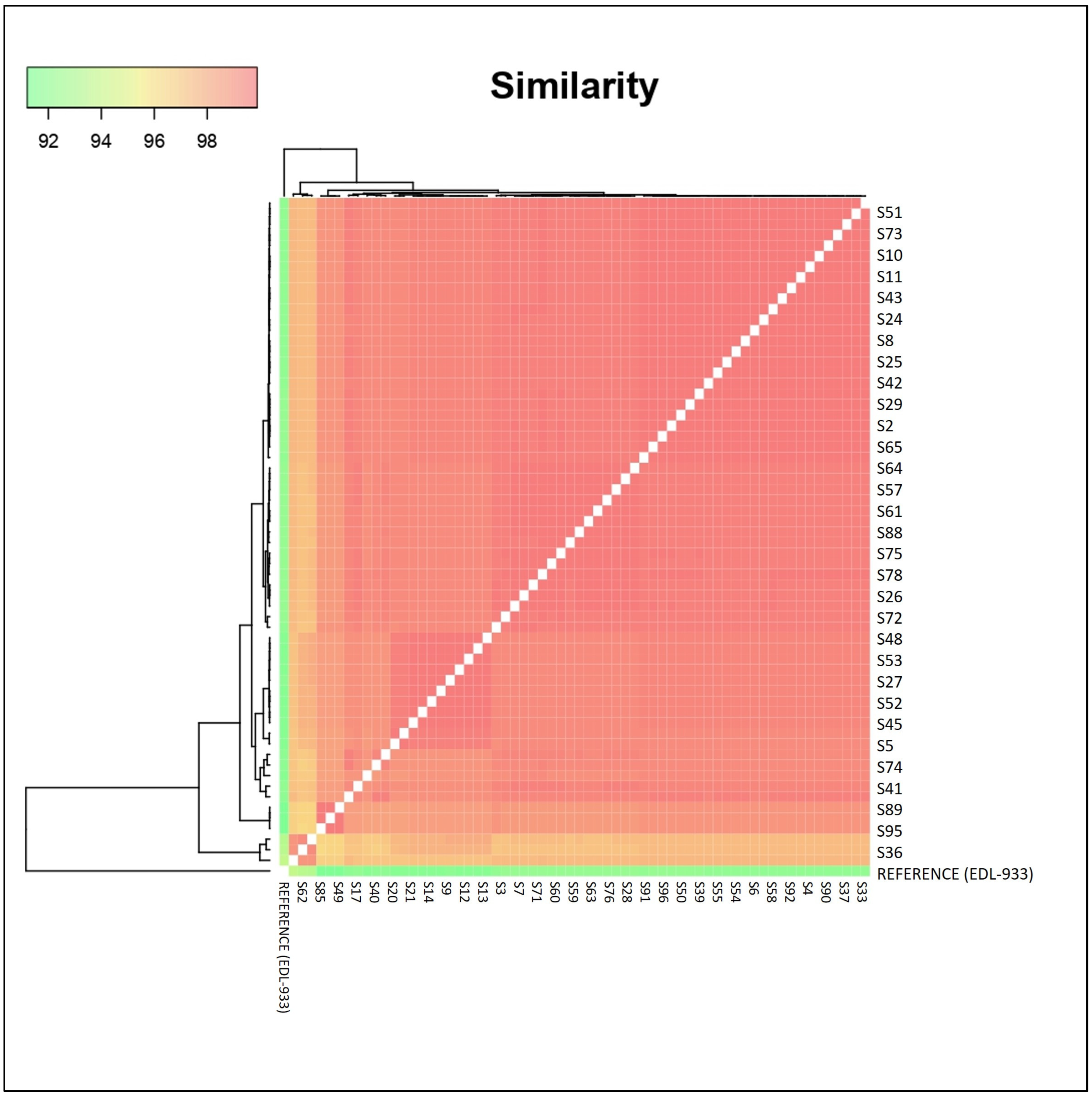
2.2. Cluster Analysis between Presumptive Super- and Low-Shedder O157 Strains
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Strains Selection, DNA Isolation, Genome Sequencing, and Quality Control
5.2. Hybrid Genome Assembly
5.3. In Silico Identification of Serogroup, Multilocus Sequence Typing (MLST), Insertion Sites, and Virulence Genes
5.4. In Silico Detection of Anti-Terminator Q933 Gene, Lineage-Specific Polymorphism Assay (LSPA-6), and Clade Genotyping using Geneious Prime
5.5. Cluster Analysis between Presumptive Super- and Low-Shedder O157 Strains
5.6. SNP Tree and Pairwise Comparison
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- AHS. E. coli Outbreak-Latest Updates. Available online: https://www.albertahealthservices.ca/topics/Page18601.aspx#sept22 (accessed on 23 September 2023).
- Kenny, B.; DeVinney, R.; Stein, M.; Reinscheid, D.J.; Frey, E.A.; Finlay, B.B. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 1997, 91, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Scheutz, F.; Teel, L.; Beutin, L.; Piérard, D.; Buvens, G.; Karch, H.; Mellmann, A.; Caprioli, A.; Tozzoli, R.; Morabito, S. Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 2012, 50, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Quiñones, B.; McMahon, S.; Mandrell, R.E. A single-step purification and molecular characterization of functional Shiga toxin 2 variants from pathogenic Escherichia coli. Toxins 2012, 4, 487–504. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.C.; Zhang, Y.; Bai, X.; Xiong, Y.; Wang, Y.; Yang, X.; Xu, Q.; He, X. Structural and functional characterization of Stx2k, a New Subtype of Shiga Toxin 2. Microorganisms 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Makino, K.; Ohnishi, M.; Kurokawa, K.; Ishii, K.; Yokoyama, K.; Han, C.-G.; Ohtsubo, E.; Nakayama, K.; Murata, T. Complete genome sequence of enterohemorrhagic Eschelichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001, 8, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A.W.; Bielaszewska, M.; Zhang, W.-L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring Shiga toxin 2 gene variants: Frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Olsen, K.E.; Ethelberg, S.; Scheutz, F. Subtyping method for Escherichia coli Shiga toxin (verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 2007, 45, 2020–2024. [Google Scholar] [CrossRef]
- Zhang, P.; Essendoubi, S.; Keenliside, J.; Reuter, T.; Stanford, K.; King, R.; Lu, P.; Yang, X. Genomic analysis of Shiga toxin-producing Escherichia coli O157:H7 from cattle and pork-production related environments. NPJ Sci. Food 2021, 5, 15. [Google Scholar] [CrossRef]
- Bumunang, E.W.; Zaheer, R.; Stanford, K.; Laing, C.; Niu, D.; Guan, L.L.; Chui, L.; Tarr, G.A.; McAllister, T.A. Genomic analysis of Shiga toxin-producing E. coli O157 cattle and clinical isolates from Alberta, Canada. Toxins 2022, 14, 603. [Google Scholar] [CrossRef]
- Heiman, K.E.; Mody, R.K.; Johnson, S.D.; Griffin, P.M.; Gould, L.H. Escherichia coli O157 outbreaks in the United States, 2003–2012. Emerg. Infect. Dis. 2015, 21, 1293. [Google Scholar] [CrossRef]
- Matthews, L.; Low, J.; Gally, D.; Pearce, M.; Mellor, D.; Heesterbeek, J.; Chase-Topping, M.; Naylor, S.; Shaw, D.; Reid, S. Heterogeneous shedding of Escherichia coli O157 in cattle and its implications for control. Proc. Natl. Acad. Sci. USA 2006, 103, 547–552. [Google Scholar] [CrossRef]
- Kempf, F.; La Ragione, R.; Chirullo, B.; Schouler, C.; Velge, P. Super shedding in enteric pathogens: A review. Microorganisms 2022, 10, 2101. [Google Scholar] [CrossRef]
- Brusa, V.; Restovich, V.; Galli, L.; Arias, R.; Linares, L.; Costa, M.; Ruiz Diaz, V.; Pugin, D.; Leotta, G. Reduction of Shiga toxin-producing Escherichia coli in a beef abattoir. Food Sci. Technol. Int. 2022, 28, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Callaway, T.R.; Eddrington, T.S.; Loneragan, G.H.; Carrr, M.A.; Nisbet, D.J. Shiga toxin producing Escherichia coli (STEC) ecology in cattle and management-based options to reduce fecal shedding. Agric. Food Analy. Bacteriol. 2013, 3, 39–69. [Google Scholar]
- Munns, K.D.; Selinger, L.B.; Stanford, K.; Guan, L.; Callaway, T.R.; McAllister, T.A. Perspectives on super-shedding of Escherichia coli O157:H7 by cattle. Foodborne Pathog. Dis. 2015, 12, 89–103. [Google Scholar] [CrossRef]
- Spencer, S.E.; Besser, T.E.; Cobbold, R.N.; French, N.P. ‘Super’ or just ‘above average’? Supershedders and the transmission of Escherichia coli O157:H7 among feedlot cattle. J. R. Soc. Interface 2015, 6, 0446. [Google Scholar] [CrossRef]
- Beauvais, W.; Gart, E.V.; Bean, M.; Blanco, A.; Wilsey, J.; McWhinney, K.; Bryan, L.; Krath, M.; Yang, C.Y.; Manriquez Alvarez, D.; et al. The prevalence of Escherichia coli O157:H7 fecal shedding in feedlot pens is affected by the water-to-cattle ratio: A randomized controlled trial. PLoS ONE 2018, 7, e0192149. [Google Scholar] [CrossRef] [PubMed]
- Venegas-Vargas, C.; Henderson, S.; Khare, A.; Mosci, R.E.; Lehnert, J.D.; Singh, P.; Ouellette, L.M.; Norby, B.; Funk, J.A.; Rust, S. Factors associated with Shiga toxin-producing Escherichia coli shedding by dairy and beef cattle. Appl. Environ. Microbiol. 2016, 82, 5049–5056. [Google Scholar] [CrossRef]
- Robinson, S.; Brown, P.; Wright, E.; Hart, C.; French, N. Quantifying within-and between-animal variation and uncertainty associated with counts of Escherichia coli O157 occurring in naturally infected cattle faeces. J. R. Soc. Interface 2009, 6, 169–177. [Google Scholar] [CrossRef]
- Ballem, A.; Goncalves, S.; Garcia-Meniño, I.; Flament-Simon, S.C.; Blanco, J.E.; Fernandes, C.; Saavedra, M.J.; Pinto, C.; Oliveira, H.; Blanco, J. Prevalence and serotypes of Shiga toxin-producing Escherichia coli (STEC) in dairy cattle from Northern Portugal. PLoS ONE 2020, 15, e0244713. [Google Scholar] [CrossRef]
- Castro, V.S.; Figueiredo, E.; McAllister, T.; Stanford, K. Farm to fork impacts of super-shedders and high-event periods on food safety. Trends Food Sci. Technol. 2022, 127, 129–142. [Google Scholar] [CrossRef]
- Williams, K.; Ward, M.; Dhungyel, O.; Hall, E. Risk factors for Escherichia coli O157 shedding and super-shedding by dairy heifers at pasture. Epidemiol. Infect. 2015, 143, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Wang, O.; McAllister, T.A.; Plastow, G.; Stanford, K.; Selinger, B.; Guan, L.L. Host mechanisms involved in cattle Escherichia coli O157 shedding: A fundamental understanding for reducing foodborne pathogen in food animal production. Sci. Rep. 2017, 7, 7630. [Google Scholar] [CrossRef] [PubMed]
- Chase-Topping, M.E.; McKendrick, I.J.; Pearce, M.C.; MacDonald, P.; Matthews, L.; Halliday, J.; Allison, L.; Fenlon, D.; Low, J.C.; Gunn, G. Risk factors for the presence of high-level shedders of Escherichia coli O157 on Scottish farms. J. Clin. Microbiol. 2007, 45, 1594–1603. [Google Scholar] [CrossRef]
- Chase-Topping, M.; Gally, D.; Low, C.; Matthews, L.; Woolhouse, M. Super-shedding and the link between human infection and livestock carriage of Escherichia coli O157. Nat. Rev. Microbiol. 2008, 6, 904–912. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Katani, R.; Cote, R.; Kudva, I.T.; DebRoy, C.; Arthur, T.M.; Kapur, V. Comparative genomics of two super-shedder isolates of Escherichia coli O157:H7. PLoS ONE 2017, 12, e0182940. [Google Scholar] [CrossRef]
- Cote, R.; Katani, R.; Moreau, M.R.; Kudva, I.T.; Arthur, T.M.; DebRoy, C.; Mwangi, M.M.; Albert, I.; Raygoza Garay, J.A.; Li, L. Comparative analysis of super-shedder strains of Escherichia coli O157:H7 reveals distinctive genomic features and a strongly aggregative adherent phenotype on bovine rectoanal junction squamous epithelial cells. PLoS ONE 2015, 10, e0116743. [Google Scholar] [CrossRef]
- Munns, K.D.; Zaheer, R.; Xu, Y.; Stanford, K.; Laing, C.R.; Gannon, V.P.; Selinger, L.B.; McAllister, T.A. Comparative genomic analysis of Escherichia coli O157:H7 isolated from super-shedder and low-shedder cattle. PLoS ONE 2016, 11, e0151673. [Google Scholar] [CrossRef]
- Yang, Z.; Kovar, J.; Kim, J.; Nietfeldt, J.; Smith, D.R.; Moxley, R.A.; Olson, M.E.; Fey, P.D.; Benson, A.K. Identification of common subpopulations of non-sorbitol-fermenting, β-glucuronidase-negative Escherichia coli O157:H7 from bovine production environments and human clinical samples. Appl. Environ. Microbiol. 2004, 70, 6846–6854. [Google Scholar] [CrossRef]
- Besser, T.E.; Shaikh, N.; Holt, N.J.; Tarr, P.I.; Konkel, M.E.; Malik-Kale, P.; Walsh, C.W.; Whittam, T.S.; Bono, J.L. Greater diversity of Shiga toxin-encoding bacteriophage insertion sites among Escherichia coli O157:H7 isolates from cattle than in those from humans. Appl. Environ. Microbiol. 2007, 73, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Manning, S.D.; Motiwala, A.S.; Springman, A.C.; Qi, W.; Lacher, D.W.; Ouellette, L.M.; Mladonicky, J.M.; Somsel, P.; Rudrik, J.T.; Dietrich, S.E. Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. USA 2008, 105, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Cowley, L.A.; McAteer, S.P.; Jenkins, C.; Dallman, T.J.; Bono, J.L.; Gally, D.L. Evolution of a zoonotic pathogen: Investigating prophage diversity in enterohaemorrhagic Escherichia coli O157 by long-read sequencing. Microb. Genom. 2016, 2, e000096. [Google Scholar] [CrossRef]
- Perna, N.T.; Plunkett, G.; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 2001, 409, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Kulasekara, B.R.; Jacobs, M.; Zhou, Y.; Wu, Z.; Sims, E.; Saenphimmachak, C.; Rohmer, L.; Ritchie, J.M.; Radey, M.; McKevitt, M. Analysis of the genome of the Escherichia coli O157:H7 2006 spinach-associated outbreak isolate indicates candidate genes that may enhance virulence. Infect. Immun. 2009, 77, 3713–3721. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Lee, S.; Park, D.; Jeong, K.C. Genetic and functional analyses of virulence potential of an Escherichia coli O157:H7 strain isolated from super-shedder cattle. Front. Cell. Infect. Microbiol. 2020, 10, 271. [Google Scholar] [CrossRef]
- Eppinger, M.; Mammel, M.K.; Leclerc, J.E.; Ravel, J.; Cebula, T.A. Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc. Natl. Acad. Sci. USA 2011, 108, 20142–20147. [Google Scholar] [CrossRef]
- Naseer, U.; Løbersli, I.; Hindrum, M.; Bruvik, T.; Brandal, L.T. Virulence factors of Shiga toxin-producing Escherichia coli and the risk of developing haemolytic uraemic syndrome in Norway, 1992–2013. Eur. J. Clin. Microbiol. 2017, 36, 1613–1620. [Google Scholar] [CrossRef]
- Byrne, L.; Dallman, T.J.; Adams, N.; Mikhail, A.F.; McCarthy, N.; Jenkins, C. Highly pathogenic clone of Shiga toxin–producing Escherichia coli O157:H7, England and Wales. Emerg. Infect. Dis. 2018, 24, 2303. [Google Scholar] [CrossRef]
- Fitzgerald, S.F.; Beckett, A.E.; Palarea-Albaladejo, J.; McAteer, S.; Shaaban, S.; Morgan, J.; Ahmad, N.I.; Young, R.; Mabbott, N.A.; Morrison, L. Shiga toxin sub-type 2a increases the efficiency of Escherichia coli O157 transmission between animals and restricts epithelial regeneration in bovine enteroids. PLoS Pathog. 2019, 15, e1008003. [Google Scholar] [CrossRef]
- Henderson, S.T.; Singh, P.; Knupp, D.; Lacher, D.W.; Abu-Ali, G.S.; Rudrik, J.T.; Manning, S.D. Variability in the occupancy of Escherichia coli O157 integration sites by Shiga toxin-encoding prophages. Toxins 2021, 13, 433. [Google Scholar] [CrossRef]
- Bielaszewska, M.; Prager, R.; Köck, R.; Mellmann, A.; Zhang, W.; Tschäpe, H.; Tarr, P.I.; Karch, H. Shiga toxin gene loss and transfer in vitro and in vivo during enterohemorrhagic Escherichia coli O26 infection in humans. Appl. Environ. Microbiol. 2007, 73, 3144–31550. [Google Scholar] [CrossRef]
- Ahmad, A.; Zurek, L. Evaluation of the anti-terminator Q933 gene as a marker for Escherichia coli O157:H7 with high Shiga toxin production. Curr. Microbiol. 2006, 53, 324–328. [Google Scholar] [CrossRef]
- Oliveira, G.S.; Lentz, S.A.M.; Wink, P.L.; Martins, A.F. Molecular typing of mcr-1 Escherichia coli isolates from pigs and farm environment based on fumC and fimH alleles. Future Microbiol. 2023, 18. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.; Agopsowicz, C.A.; McAllister, T.A. Genetic diversity and antimicrobial resistance among isolates of Escherichia coli O157:H7 from feces and hides of super-shedders and low-shedding pen-mates in two commercial beef feedlots. BMC Vet. Res. 2012, 8, 178. [Google Scholar] [CrossRef]
- Pianciola, L.; Rivas, M. Genotypic features of clinical and bovine Escherichia coli O157 strains isolated in countries with different associated-disease incidences. Microorganisms 2018, 6, 36. [Google Scholar] [CrossRef]
- Sharma, R.; Stanford, K.; Louie, M.; Munns, K.; John, S.J.; Zhang, Y.; Gannon, V.; Chui, L.; Read, R.; Topp, E. Escherichia coli O157:H7 lineages in healthy beef and dairy cattle and clinical human cases in Alberta, Canada. J. Food Prot. 2009, 72, 601–607. [Google Scholar] [CrossRef]
- Ogura, Y.; Mondal, S.I.; Islam, M.R.; Mako, T.; Arisawa, K.; Katsura, K.; Ooka, T.; Gotoh, Y.; Murase, K.; Ohnishi, M. The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157:H7 is correlated with the subtypes of toxin-encoding phage. Sci. Rep. 2015, 5, 16663. [Google Scholar] [CrossRef] [PubMed]
- Yara, D.A.; Greig, D.R.; Gally, D.L.; Dallman, T.J.; Jenkins, C. Comparison of Shiga toxin-encoding bacteriophages in highly pathogenic strains of Shiga toxin-producing Escherichia coli O157:H7 in the UK. Microb. Genom. 2020, 6, e000334. [Google Scholar] [CrossRef] [PubMed]
- Dewell, G.A.; Simpson, C.A.; Dewell, R.D.; Hyatt, D.R.; Belk, K.E.; Scanga, J.A.; Morley, P.S.; Grandin, T.; Smith, G.C.; Dargatz, D.A.; et al. Impact of transportation and lairage on hide contamination with Escherichia coli O157 in finished beef cattle. J. Food Protect. 2008, 71, 1114–1118. [Google Scholar] [CrossRef]
- Browne, A.S.; Midwinter, A.C.; Withers, H.; Cookson, A.L.; Biggs, P.J.; Marshall, J.C.; Benschop, J.; Hathaway, S.; Rogers, L.; Nisa, S. Transmission dynamics of Shiga toxin-producing Escherichia coli in New Zealand cattle from farm to slaughter. Appl. Environ. Microbiol. 2021, 87, e02907–e02920. [Google Scholar] [CrossRef] [PubMed]
- Stephens, T.; McAllister, T.; Stanford, K. Perineal swabs reveal effect of super shedders on the transmission of Escherichia coli O157:H7 in commercial feedlots. J. Anim. Sci. 2009, 87, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Stanford, K.; Hannon, S.; Booker, C.W.; Jim, G.K. Variable efficacy of a vaccine and direct-fed microbial for controlling Escherichia coli O157:H7 in feces and on hides of feedlot cattle. Foodborne Pathog. Dis. 2014, 11, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P. metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Ingle, D.J.; Valcanis, M.; Kuzevski, A.; Tauschek, M.; Inouye, M.; Stinear, T.; Levine, M.M.; Robins-Browne, R.M.; Holt, K.E. In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microb. Genom. 2016, 2, e000064. [Google Scholar] [CrossRef]
- Bessonov, K.; Laing, C.; Robertson, J.; Yong, I.; Ziebell, K.; Gannon, V.P.; Nichani, A.; Arya, G.; Nash, J.H.; Christianson, S. ECTyper: In silico Escherichia coli serotype and species prediction from raw and assembled whole-genome sequence data. Microb. Genom. 2021, 7, 000728. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef]
- Malberg Tetzschner, A.M.; Johnson, J.R.; Johnston, B.D.; Lund, O.; Scheutz, F. In silico genotyping of Escherichia coli isolates for extraintestinal virulence genes by use of whole-genome sequencing data. J. Clin. Microbiol. 2020, 58, e01269-20. [Google Scholar] [CrossRef]
- Hartzell, A.; Chen, C.; Lewis, C.; Liu, K.; Reynolds, S.; Dudley, E.G. Escherichia coli O157:H7 of genotype lineage-specific polymorphism assay 211111 and clade 8 are common clinical isolates within Pennsylvania. Foodborne Pathog. Dis. 2011, 8, 763–768. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Isolates | Source | MLST | Lineage | Clade | Insertion Site | stx Subtype | eae | tir | ehxA | Q933 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 33 | SS | 11 | I | 1 | mlrA | wrbA | Stx1a/2a | + | + | + | + |
| 1 | SS | 8 | mlrA | wrbA | Stx 1a/2a | + | + | + | + | ||
| 1 | SS | 1 | mlrA | - | Stx 1a | + | + | + | + | ||
| 24 | LS | 1 | mlrA | wrbA | Stx 1a/2a | + | + | + | + | ||
| 1 | LS | 8 | mlrA | wrbA | Stx 1a/2a | + | + | + | + | ||
| 1 | LS | 1 | mlrA | - | Stx 1a | + | + | + | + | ||
| 1 | LS | 1 | mlrA * | wrbA | Stx 2a | + | + | + | + | ||
| 1 | LS | 1 | mlrA | wrbA * | Stx 1a | + | + | + | + | ||
| Strain ID | Source | Lineage | Clade | stx Subtype | Insertion Site | Accession Number | Year Isolated | References | |
|---|---|---|---|---|---|---|---|---|---|
| Sakai | Clinical | I | 1 | Stx1a/2a | mlrA | wrbA | NC002695.2 | 1996 | [6] |
| EDL933 | Ground beef | I | 3 | Stx1a/2a | mlrA | wrbA | CP008957.1 | 1982 | [35] |
| TW14359 | Clinical | I/II | 8 | Stx2a/2c | - | argW | NC013008.1 | 2006 | [36] |
| JEONG-1266 | Cattle | I/II | 8 | Stx2a/2c | - | argW | CP014314 | 2011 | [37] |
| SS17 | Cattle | I/II | 8 | Stx2a/2c | - | argW | CP008805.1 | 2009/10 | [29] |
| SS52 | Cattle | I/II | - | - | - | - | CP010304.1 | 2009/10 | [28] |
| EC4115.1 | Spinach | I/II | 8 | Stx2a/2c | - | argW | NC011353.1 | 2006 | [38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bumunang, E.W.; Castro, V.S.; Alexander, T.; Zaheer, R.; McAllister, T.A.; Guan, L.L.; Stanford, K. In Silico Analysis of Shiga Toxin-Producing Escherichia coli O157:H7 Strains from Presumptive Super- and Low-Shedder Cattle. Toxins 2024, 16, 86. https://doi.org/10.3390/toxins16020086
Bumunang EW, Castro VS, Alexander T, Zaheer R, McAllister TA, Guan LL, Stanford K. In Silico Analysis of Shiga Toxin-Producing Escherichia coli O157:H7 Strains from Presumptive Super- and Low-Shedder Cattle. Toxins. 2024; 16(2):86. https://doi.org/10.3390/toxins16020086
Chicago/Turabian StyleBumunang, Emmanuel W., Vinicius S. Castro, Trevor Alexander, Rahat Zaheer, Tim A. McAllister, Le Luo Guan, and Kim Stanford. 2024. "In Silico Analysis of Shiga Toxin-Producing Escherichia coli O157:H7 Strains from Presumptive Super- and Low-Shedder Cattle" Toxins 16, no. 2: 86. https://doi.org/10.3390/toxins16020086
APA StyleBumunang, E. W., Castro, V. S., Alexander, T., Zaheer, R., McAllister, T. A., Guan, L. L., & Stanford, K. (2024). In Silico Analysis of Shiga Toxin-Producing Escherichia coli O157:H7 Strains from Presumptive Super- and Low-Shedder Cattle. Toxins, 16(2), 86. https://doi.org/10.3390/toxins16020086