Effects of Deoxynivalenol and Its Acetylated Derivatives on Lipid Metabolism in Human Normal Hepatocytes

 , and
, and
Abstract
:1. Introduction
2. Results
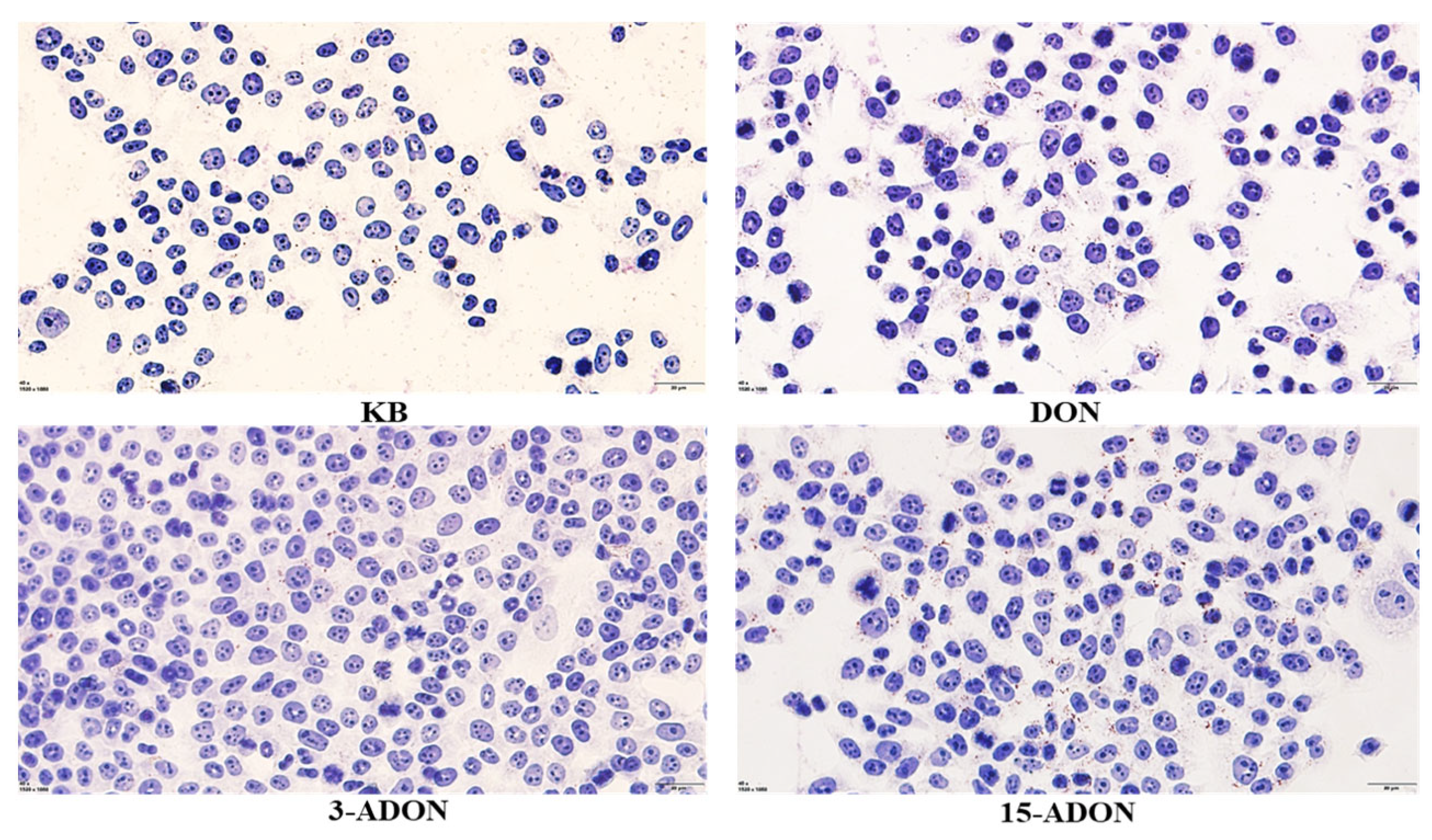
2.1. Effect of DON and Its Acetylated Derivatives on Cellular Activity and Lipid Droplet Accumulation
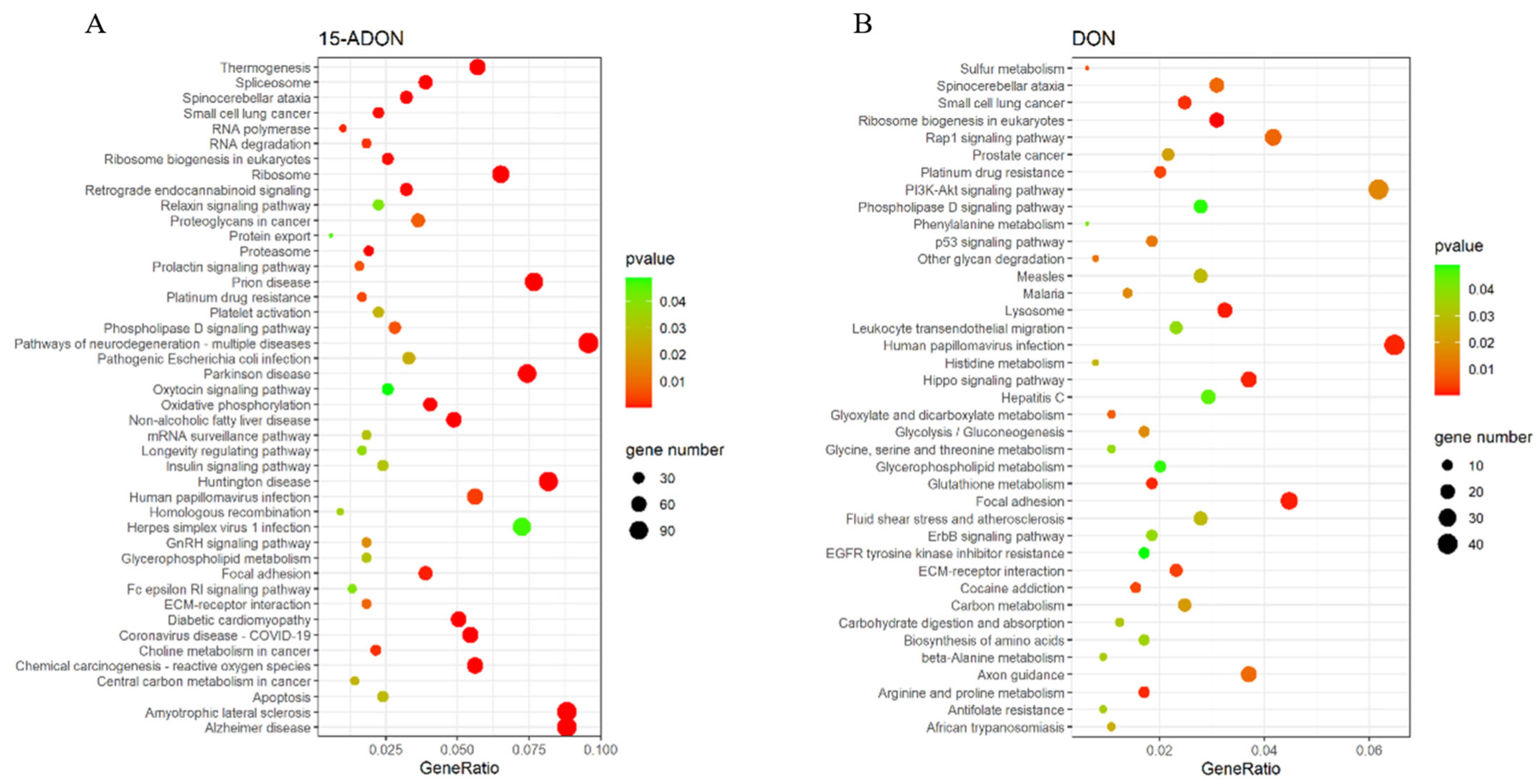
2.2. DON and Its Acetylated Derivatives on the Transcriptome Effects
2.3. DON and Its Acetylated Derivatives on L-02 Cells Effects of Lipidomics
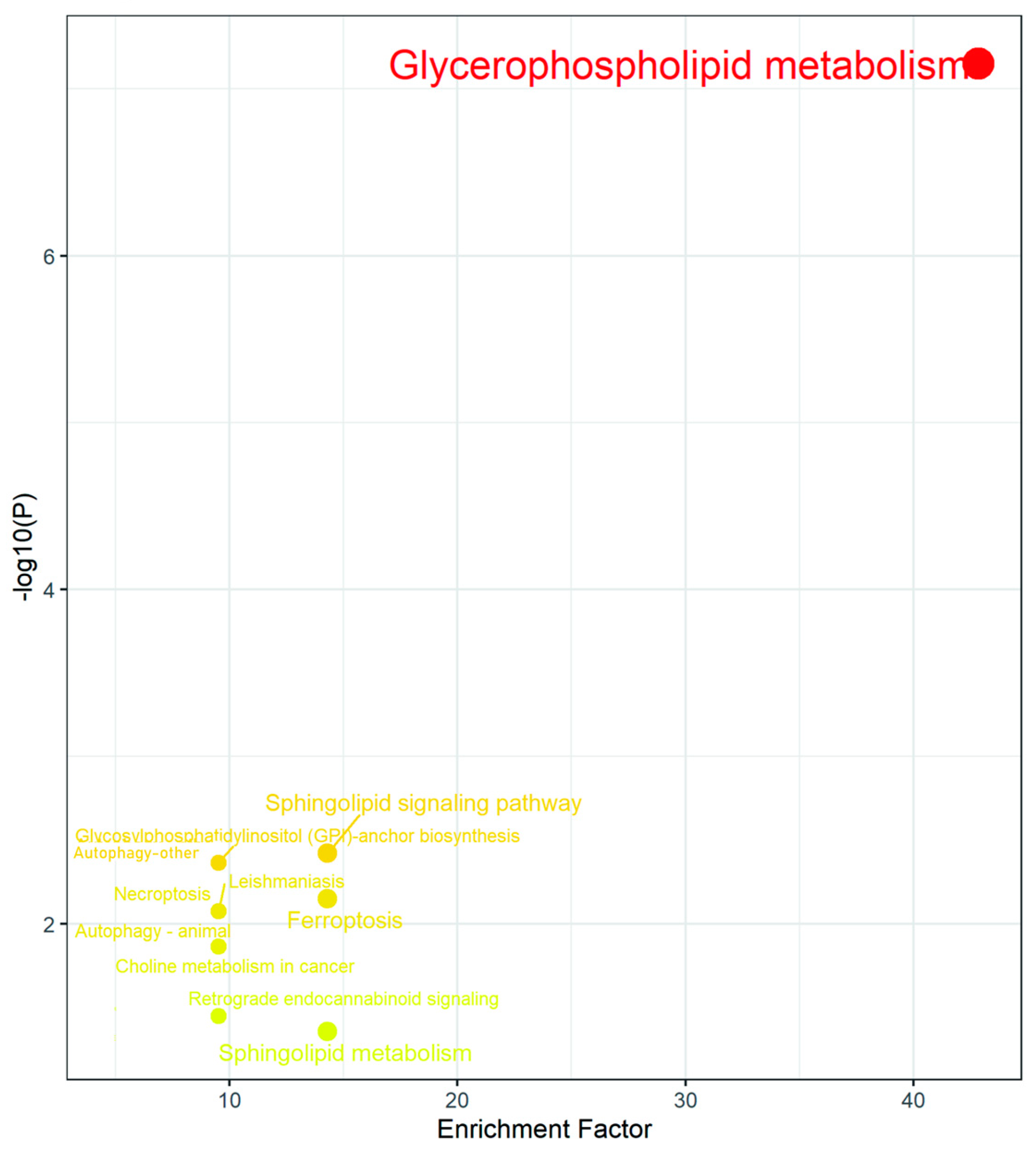
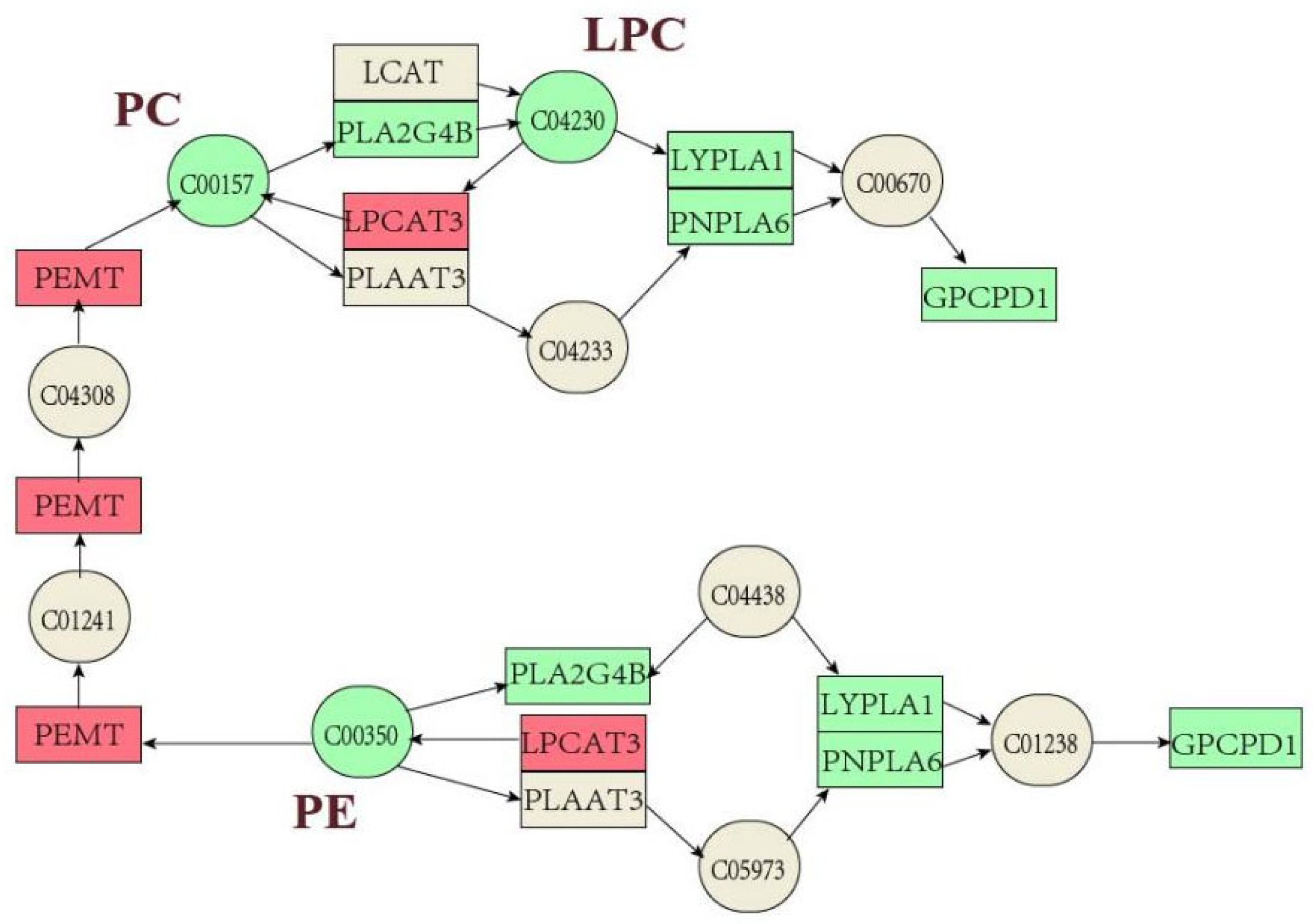
2.4. Combined Transcriptomics and Lipidomics Analysis
3. Disscusion
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. Cell Culture
5.3. Cytotoxicity Assays
5.4. Oil Red O Staining
5.5. RNA Extraction
5.6. RNA-Seq Analysis
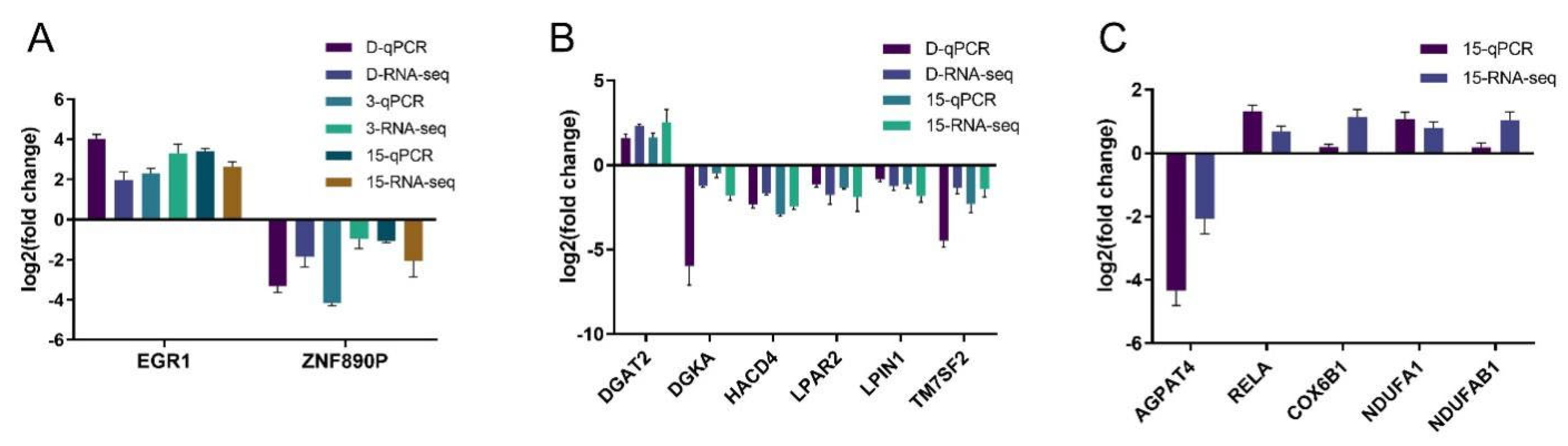
5.7. Quantitative Real-Time PCR (qRT-PCR)
5.8. Metabolomic Analysis
5.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- EFSA Panel on Contaminants in the Food Chain. Scientific Opinion on the risks for human and animal health related to the presence of modified forms of certain mycotoxins in food and feed. EFSA J. 2014, 12, 3916. [Google Scholar]
- Khaneghah, A.M.; Fakhri, Y.; Raeisi, S.; Armoon, B.; Sant’Ana, A.S. Prevalence and concentration of ochratoxin A, zearalenone, deoxynivalenol and total aflatoxin in cereal-based products: A systematic review and meta-analysis. Food Chem. Toxicol. 2018, 118, 830–848. [Google Scholar] [CrossRef]
- Alizadeh, A.; Braber, S.; Akbari, P.; Kraneveld, A.; Garssen, J.; Fink-Gremmels, J. Deoxynivalenol and its modified forms: Are there major differences? Toxins 2016, 8, 334. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; Xu, J.; Liu, X.; Yin, X.; Shi, J. Natural occurrence of deoxynivalenol and zearalenone in wheat from Jiangsu province, China. Food Chem. 2014, 157, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Joint FAO/WHO Expert Committee on Food Additives; Food and Agriculture Organization of the United Nations; World Health Organization. Evaluation of Certain Contaminants in Food: Seventy-Second Report of the Joint FAO/WHO Expert Committe on Food Additives; World Health Organization: Geneva, Switzerland, 2011; 105p. [Google Scholar]
- EFSA Panel on Contaminants in the Food Chain; Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Grasl-Kraupp, B. Risks to human and animal health related to the presence of deoxynivalenol and its acetylated and modified forms in food and feed. EFSA J. 2017, 15, e04718. [Google Scholar] [PubMed]
- De Boevre, M.; Di Mavungu, J.D.; Maene, P.; Audenaert, K.; Deforce, D.; Haesaert, G.; Eeckhout, M.; Callebaut, A.; Berthiller, F.; Van Peteghem, C. Development and validation of an LC-MS/MS method for the simultaneous determination of deoxynivalenol, zearalenone, T-2-toxin and some masked metabolites in different cereals and cereal-derived food. Food Addit. Contam. Part A 2012, 29, 819–835. [Google Scholar] [CrossRef]
- Palacios, S.A.; Erazo, J.G.; Ciasca, B.; Lattanzio, V.M.; Reynoso, M.M.; Farnochi, M.C.; Torres, A.M. Occurrence of deoxynivalenol and deoxynivalenol-3-glucoside in durum wheat from Argentina. Food Chem. 2017, 230, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Carballo, D.; Moltó, J.; Berrada, H.; Ferrer, E. Presence of mycotoxins in ready-to-eat food and subsequent risk assessment. Food Chem. Toxicol. 2018, 121, 558–565. [Google Scholar] [CrossRef]
- Broekaert, N.; Devreese, M.; De Mil, T.; Fraeyman, S.; Antonissen, G.; De Baere, S.; De Backer, P.; Vermeulen, A.; Croubels, S. Oral bioavailability, hydrolysis, and comparative toxicokinetics of 3-acetyldeoxynivalenol and 15-acetyldeoxynivalenol in broiler chickens and pigs. J. Agric. Food Chem. 2015, 63, 8734–8742. [Google Scholar] [CrossRef]
- Kadota, T.; Furusawa, H.; Hirano, S.; Tajima, O.; Kamata, Y.; Sugita-Konishi, Y. Comparative study of deoxynivalenol, 3-acetyldeoxynivalenol, and 15-acetyldeoxynivalenol on intestinal transport and IL-8 secretion in the human cell line Caco-2. Toxicol. In Vitro 2013, 27, 1888–1895. [Google Scholar] [CrossRef]
- Hänninen, O.; Lindström-Seppä, P.; Pelkonen, K. Role of gut in xenobiotic metabolism. Arch. Toxicol. 1987, 60, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, M.J.; Tsurkan, L.; Garrett, M.; Shaver, T.M.; Hyatt, J.L.; Edwards, C.C.; Hicks, L.D.; Potter, P.M. Organ-specific carboxylesterase profiling identifies the small intestine and kidney as major contributors of activation of the anticancer prodrug CPT-11. Biochem. Pharmacol. 2011, 81, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, C.; Airault, C.; Djelloul, M.; Guillebaud, F.; Barbouche, R.; Troadec, J.-D.; Gaigé, S.; Dallaporta, M. The food born mycotoxin deoxynivalenol induces low-grade inflammation in mice in the absence of observed-adverse effects. Toxicol. Lett. 2015, 232, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yu, M.; Wu, Q.; Peng, Z.; Wang, D.; Kuča, K.; Yao, P.; Yan, H.; Nüssler, A.K.; Liu, L. Gender and geographical variability in the exposure pattern and metabolism of deoxynivalenol in humans: A review. J. Appl. Toxicol. 2017, 37, 60–70. [Google Scholar] [CrossRef]
- Pietsch, C.; Schulz, C.; Rovira, P.; Kloas, W.; Burkhardt-Holm, P. Organ damage and hepatic lipid accumulation in carp (Cyprinus carpio L.) after feed-borne exposure to the mycotoxin, deoxynivalenol (DON). Toxins 2014, 6, 756–778. [Google Scholar] [CrossRef]
- Abdel-Wahhab, M.A.; El-Kady, A.A.; Hassan, A.M.; Abd El-Moneim, O.M.; Abdel-Aziem, S.H. Effectiveness of activated carbon and Egyptian montmorillonite in the protection against deoxynivalenol-induced cytotoxicity and genotoxicity in rats. Food Chem. Toxicol. 2015, 83, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Barbouche, R.; Gaigé, S.; Airault, C.; Poirot, K.; Dallaporta, M.; Troadec, J.-D.; Abysique, A. The food contaminant deoxynivalenol provokes metabolic impairments resulting in non-alcoholic fatty liver (NAFL) in mice. Sci. Rep. 2020, 10, 12072. [Google Scholar] [CrossRef] [PubMed]
- Alassane-Kpembi, I.; Pinton, P.; Hupé, J.-F.; Neves, M.; Lippi, Y.; Combes, S.; Castex, M.; Oswald, I.P. Saccharomyces cerevisiae boulardii reduces the deoxynivalenol-induced alteration of the intestinal transcriptome. Toxins 2018, 10, 199. [Google Scholar] [CrossRef]
- Peng, Z.; Chen, L.; Nüssler, A.K.; Liu, L.; Yang, W. Current sights for mechanisms of deoxynivalenol-induced hepatotoxicity and prospective views for future scientific research: A mini review. J. Appl. Toxicol. 2017, 37, 518–529. [Google Scholar] [CrossRef]
- Sun, L.-H.; Lei, M.-y.; Zhang, N.-Y.; Gao, X.; Li, C.; Krumm, C.S.; Qi, D.-S. Individual and combined cytotoxic effects of aflatoxin B1, zearalenone, deoxynivalenol and fumonisin B1 on BRL 3A rat liver cells. Toxicon 2015, 95, 6–12. [Google Scholar] [CrossRef]
- He, Y.; Yin, X.; Dong, J.; Yang, Q.; Wu, Y.; Gong, Z. Transcriptome analysis of Caco-2 cells upon the exposure of mycotoxin deoxynivalenol and its acetylated derivatives. Toxins 2021, 13, 167. [Google Scholar] [CrossRef]
- Desjardins, A.E.; McCormick, S.P.; Appell, M. Structure-activity relationships of trichothecene toxins in an Arabidopsis thaliana leaf assay. J. Agric. Food Chem. 2007, 55, 6487–6492. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.L.; Wannemacher, R.W., Jr. Structure-function relationships of 12, 13-epoxytrichothecene mycotoxins in cell culture: Comparison to whole animal lethality. Toxicon 1986, 24, 985–994. [Google Scholar] [CrossRef]
- Zhou, C.; Jia, H.-m.; Liu, Y.-t.; Yu, M.; Chang, X.; Ba, Y.-m.; Zou, Z.-m. Metabolism of glycerophospholipid, bile acid and retinol is correlated with the early outcomes of autoimmune hepatitis. Mol. BioSyst. 2016, 12, 1574–1585. [Google Scholar] [CrossRef]
- Alassane-Kpembi, I.; Canlet, C.; Tremblay-Franco, M.; Jourdan, F.; Chalzaviel, M.; Pinton, P.; Cossalter, A.M.; Achard, C.; Castex, M.; Combes, S. 1H-NMR metabolomics response to a realistic diet contamination with the mycotoxin deoxynivalenol: Effect of probiotics supplementation. Food Chem. Toxicol. 2020, 138, 111222. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Kawai, D.; Yamamoto, K. Molecular mechanisms and new treatment strategies for non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2014, 15, 7352–7379. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, Z.; Yang, Z.; Qi, Y.; Shi, H.; Zhou, Y.; Wang, F.; Yang, M. Serum metabolomic profiling reveals an increase in homocitrulline in Chinese patients with nonalcoholic fatty liver disease: A retrospective study. PeerJ 2021, 9, e11346. [Google Scholar] [CrossRef]
- Wang, X.; Wu, L.; Tao, J.; Ye, H.; Wang, J.; Gao, R.; Liu, W. A lipidomic approach to bisphenol F-induced non-alcoholic fatty liver disease-like changes: Altered lipid components in a murine model. Environ. Sci. Pollut. Res. 2023, 30, 112644–112659. [Google Scholar] [CrossRef]
- Sun, C.; Guo, Y.; Cong, P.; Tian, Y.; Gao, X. Liver Lipidomics Analysis Revealed the Novel Ameliorative Mechanisms of L-Carnitine on High-Fat Diet-Induced NAFLD Mice. Nutrients 2023, 15, 1359. [Google Scholar] [CrossRef]
- Feng, S.; Gan, L.; Yang, C.S.; Liu, A.B.; Lu, W.; Shao, P.; Dai, Z.; Sun, P.; Luo, Z. Effects of stigmasterol and β-sitosterol on nonalcoholic fatty liver disease in a mouse model: A lipidomic analysis. J. Agric. Food Chem. 2018, 66, 3417–3425. [Google Scholar] [CrossRef]
- Rodriguez-Cuenca, S.; Pellegrinelli, V.; Campbell, M.; Oresic, M.; Vidal-Puig, A. Sphingolipids and glycerophospholipids–the “ying and yang” of lipotoxicity in metabolic diseases. Prog. Lipid Res. 2017, 66, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Zia, Y.; Al Rajabi, A.; Mi, S.; Ju, T.; Leonard, K.-A.; Nelson, R.; Thiesen, A.; Willing, B.P.; Field, C.J.; Curtis, J.M. Hepatic expression of PEMT, but not dietary choline supplementation, reverses the protection against atherosclerosis in Pemt−/−/Ldlr−/− mice. J. Nutr. 2018, 148, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Arendt, B.M.; Ma, D.W.; Simons, B.; Noureldin, S.A.; Therapondos, G.; Guindi, M.; Sherman, M.; Allard, J.P. Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl. Physiol. Nutr. Metab. 2013, 38, 334–340. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, J.N.; Lingrell, S.; Gao, X.; Quiroga, A.D.; Takawale, A.; Armstrong, E.A.; Yager, J.Y.; Kassiri, Z.; Lehner, R.; Vance, D.E. Pioglitazone attenuates hepatic inflammation and fibrosis in phosphatidylethanolamine N-methyltransferase-deficient mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 310, G526–G538. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Da Costa, K.A.; Fischer, L.M.; Kohlmeier, M.; Kwock, L.; Wang, S.; Zeisel, S.H. Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD). FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1266–1271. [Google Scholar]
- Presa, N.; Clugston, R.D.; Lingrell, S.; Kelly, S.E.; Merrill, A.H., Jr.; Jana, S.; Kassiri, Z.; Gómez-Muñoz, A.; Vance, D.E.; Jacobs, R.L. Vitamin E alleviates non-alcoholic fatty liver disease in phosphatidylethanolamine N-methyltransferase deficient mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Kondo, T.; Sajan, M.; Luo, J.; Bronson, R.; Asano, T.; Farese, R.; Cantley, L.C.; Kahn, C.R. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKCλ/ζ. Cell Metab. 2006, 3, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Xiong, Y.; Borck, P.C.; Jang, C.; Doulias, P.-T.; Papazyan, R.; Fang, B.; Jiang, C.; Zhang, Y.; Briggs, E.R. Diet-induced circadian enhancer remodeling synchronizes opposing hepatic lipid metabolic processes. Cell 2018, 174, 831–842.e12. [Google Scholar] [CrossRef] [PubMed]
- Hodel, C. Myopathy and rhabdomyolysis with lipid-lowering drugs. Toxicol. Lett. 2002, 128, 159–168. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Boergesen, M.; Pedersen, T.Å.; Gross, B.; van Heeringen, S.J.; Hagenbeek, D.; Bindesbøll, C.; Caron, S.; Lalloyer, F.; Steffensen, K.R.; Nebb, H.I. Genome-wide profiling of liver X receptor, retinoid X receptor, and peroxisome proliferator-activated receptor α in mouse liver reveals extensive sharing of binding sites. Mol. Cell. Biol. 2012, 32, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Sun, Q.; Xu, H.; Yu, X.; Chen, W.; Wei, H.; Jiang, J.; Xu, Y.; Lu, W. Hyperuricemia induces lipid disturbances mediated by LPCAT3 upregulation in the liver. FASEB J. 2020, 34, 13474–13493. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Wang, B.; Dunham, M.M.; Hedde, P.N.; Wong, J.S.; Gratton, E.; Young, S.G.; Ford, D.A.; Tontonoz, P. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. eLife 2015, 4, e06557. [Google Scholar] [CrossRef] [PubMed]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ridgway, N.D. Phosphatidylcholine synthesis regulates triglyceride storage and chylomicron secretion by Caco2 cells. J. Lipid Res. 2018, 59, 1940–1950. [Google Scholar] [CrossRef]
- Gant, T.W.; Sauer, U.G.; Zhang, S.D.; Chorley, B.N.; Hackermüller, J.; Perdichizzi, S.; Tollefsen, K.E.; van Ravenzwaay, B.; Yauk, C.; Tong, W.; et al. A generic transcriptomics reporting framework (TRF) for ‘omics data processing and analysis. Regul. Toxicol. Pharmacol. 2017, 91, S36–S45. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments; Oxford University Press: Oxford, UK, 2009. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Pathway | Gene | |
|---|---|---|---|
| DON | has 00564 | Glycerophospholipid metabolism | LPCAT3/MBOAT2/PNPLA6/DGKA/LPIN1/PLD3/PLA2G15/DGKQ/ETNK2/PLD2/LPCAT4/PNPLA7/PLD4 |
| has 04072 | Phospholipase D signaling pathway | PLCB2/RALGDS/PLCB3/DGKA/PDGFB/CYTH2/PLCG1/DGKQ/LPAR5/PLD2/LPAR2/PTK2B/PIK3R2/DNM3/RAPGEF4/PIK3CD/SHC3/PDGFRB | |
| 15-ADON | has 04932 | Non-alcoholic fatty liver disease | SDHB/NDUFS5/JUN/AKT3/NDUFB8/CYP2E1/NDUFS3/COX8A/RELA/NDUFS8/NDUFC2-KCTD14/NDUFC2/TNFRSF1A/NDUFA4L2/NDUFA12/COX6A1/EIF2S1/FOS/NDUFB1/NDUFB10/NDUFAB1/COX4I1/SOCS3/NDUFV2/UQCR11/NDUFA11/NDUFA7/NDUFB7/PIK3R2/NDUFA13/UQCRFS1/COX6B1/GSK3A/NDUFA3/COX5B/NDUFB3/IRS1/UQCR10/NDUFA6/PPARA/NDUFB4/PIK3CA/NDUFS6/NDUFS4/PIK3R1/COX7C/UQCRQ/NDUFA2/MAPK13/CYCS/MLXIPL/NDUFB2/COX6C/NDUFB9/CYC1/NDUFA8/NDUFB11/COX7B/NDUFA1 |
| has 04072 | Phospholipase D signaling pathway | DNM3/ARF1/AKT3/HRAS/GAB2/DGKA/KITLG/DGKH/SOS2/PLCB2/JMJD7-PLA2G4B/PLA2G4B/PLD2/GRB2/PIK3R2/LPAR2/CYTH2/SOS1/RAPGEF4/PLCB1/PLCG1/PLD1/PIK3CA/DGKQ/PIK3R1/F2R/PDGFRB/AGPAT4/CYTH3/DGKI/PTK2B/SHC3/TSC1/RALGDS | |
| has 00564 | Glycerophospholipid metabolism | LPCAT3/GPD1/DGKA/DGKH/PLD4/LPCAT4/JMJD7-PLA2G4B/PLA 2G4B/PLD2/PEMT/LPIN2/MBOAT2/LPIN1/GPCPD1/PLA2G6/CHKB/PLD1/DGKQ/MBOAT1/AGPAT4/DGKI/PNPLA7 |
| Ion Mode | Group | R2Y | Q2 |
|---|---|---|---|
| Postive | DON | 0.967 | 0.704 |
| 3-ADON | 0.808 | 0.550 | |
| 15-ADON | 0.513 | 0.501 | |
| Negtive | DON | 0.989 | 0.928 |
| 3-ADON | 0.825 | 0.557 | |
| 15-ADON | 0.986 | 0.927 |
| Pathway Name | p-Value | Adj. p-Value |
|---|---|---|
| Glycerophospholipid metabolism | 3.22 × 10−7 | 8.7 × 10−6 |
| Sphingolipid signaling pathway | 0.004887 | 0.033762 |
| Glycosylphosphatidylinositol (GPI) | 0.005136 | 0.033762 |
| Autophagy—other | 0.005136 | 0.033762 |
| Ferroptosis | 0.009069 | 0.033762 |
| Compound Number | Summation Ions | Compound Name | p-Value | 15A | 3A | DON | |
|---|---|---|---|---|---|---|---|
| LIPIDMAPS | HMDB | log2(FC) | log2(FC) | log2(FC) | |||
| LMGP01010566 | HMDB0007969 | [M + HCOO]− | PC 16:0/16:1 | 3.31 × 10−7 | −1.31953 | −0.25847 | −1.62121 |
| LMGP01010535 | HMDB0007936 | [M + HCOO]− | PC 15:0/16:1 | 3.40 × 10−5 | −1.38383 | −0.40766 | −1.25755 |
| LMGP01011485 | HMDB0008008 | [M + HCOO]− | PC 16:1/18:3 | 3.46 × 10−5 | −1.58501 | −0.0225 | −1.98641 |
| LMGP01010688 | HMDB0008005 | [M + HCOO]− | PC 16:1/18:1 | 5.38 × 10−5 | −0.83986 | −0.00508 | −2.15982 |
| LMGP01010564 | HMDB0000564 | [M + H]+ | PC 16:0/16:0 | 5.79 × 10−5 | −1.23797 | −0.52877 | −1.17285 |
| LMGP02010302 | HMDB0008824 | [M − H]− | PE 14:0/16:0 | 0.000104 | −1.02936 | −0.0167 | −1.03175 |
| LMGP01010492 | HMDB0007873 | [M + H]+ | PC 14:0/18:1 | 0.000106 | −1.53803 | −0.27126 | −1.1184 |
| LMGP01010690 | HMDB0008006 | [M + H]+ | PC 16:1/18:2 | 0.000203 | −1.15744 | −0.00891 | −1.4226 |
| LMGP01010496 | HMDB0007874 | [M + HCOO]− | PC 14:0/18:2 | 0.000456 | −0.68962 | −0.39123 | −0.53687 |
| LMGP01010496 | HMDB0007874 | [M + HCOO]− | PC 16:1/16:1 | 0.002201 | −0.68962 | −0.39123 | −0.53687 |
| LMGP01010913 | HMDB0008123 | [M + H]+ | PC 18:1/22:6 | 0.009175 | −0.75511 | −0.31468 | −0.50407 |
| LMGP01012166 | HMDB0008118 | [M + H]+ | PC 18:1/22:1 | 0.009942 | −0.90162 | −0.22761 | −0.6374 |
| LMGP01010535 | HMDB0007936 | [M + H]+ | PC 15:0/16:1 | 0.02051 | −0.74051 | −0.26041 | −0.20947 |
| LMGP01010532 | HMDB0007935 | [M + H]+ | PC 15:0/16:0 | 0.022227 | −0.62339 | −0.18916 | −0.31691 |
| LMGP02011197 | HMDB0009062 | [M + H]+ | PE 18:1/18:3 | 0.026896 | −0.55402 | −0.01663 | −0.29485 |
| Primer Name | F | R |
|---|---|---|
| ß-actin | CCTTCCTGGGCATGGAGTC | TGATCTTCATTGTGCTGGGTG |
| AGPAT4 | GCTTTGGGCTGTTAGGGGGCTC | AGGAAAAAATACTTCTCGGGGT |
| DGAT2 | GAACCGCAAGGGCTTTGTGAAA | GCATGGGGCGAAACCAATGTAT |
| DGKA | ATGTTCCTGATAGCCGGATTTT | AAGGGGACTGGGTCACTTTTTT |
| HACD4 | CTTTGCCAATCCGTATCTCTCC | CCCTCTTCAACTTTGCCTCTTC |
| LPAR2 | GCCACCCCCGCTACCGAGAGAC | CAAGAGTACACAGCAGCATTGA |
| TM7SF2 | GGCAGGAATTGAAGGACAAGAG | CGAAGAAACAGATACGAGGGTT |
| RELA | AGAAGAGCAGCGTGGGGACTAC | TGCACATCAGCTTGCGAAAAGG |
| COX6B1 | GCTTCCCCAACCAGAACCAGAC | TCAGCCCGTTGCTCATCCCAGT |
| NDUFA1 | ATGTGGTTCGAGATTCTCCCCG | TCTTTCCATCAGACTCCAGTGA |
| NDUFAB1 | CCTCTCAGCACCGCTCTCTGCT | TCCTGGATGCCCTCTAACGTCA |
| EGR1 | TCTGACCCGTTCGGATCCTTTC | CTCCACCAGCACCTTCTCGTTG |
| ZNF890P | CAGGGGTCACTGTCATTCGGGG | TTCGTGTTGCTGTTGGCTATCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Z.; He, Y.; Li, Y.; Wang, Q.; Fang, M.; Yang, Q.; Gong, Z.; Xu, L. Effects of Deoxynivalenol and Its Acetylated Derivatives on Lipid Metabolism in Human Normal Hepatocytes. Toxins 2024, 16, 294. https://doi.org/10.3390/toxins16070294
Ma Z, He Y, Li Y, Wang Q, Fang M, Yang Q, Gong Z, Xu L. Effects of Deoxynivalenol and Its Acetylated Derivatives on Lipid Metabolism in Human Normal Hepatocytes. Toxins. 2024; 16(7):294. https://doi.org/10.3390/toxins16070294
Chicago/Turabian StyleMa, Zhaoqing, Yuyun He, Yuzhi Li, Qiao Wang, Min Fang, Qing Yang, Zhiyong Gong, and Lin Xu. 2024. "Effects of Deoxynivalenol and Its Acetylated Derivatives on Lipid Metabolism in Human Normal Hepatocytes" Toxins 16, no. 7: 294. https://doi.org/10.3390/toxins16070294