Identification of a Chimera Mass Spectrum of Isomeric Lipid A Species Using Negative Ion Tandem Mass Spectrometry


Abstract
:
1. Introduction
2. Results and Discussion
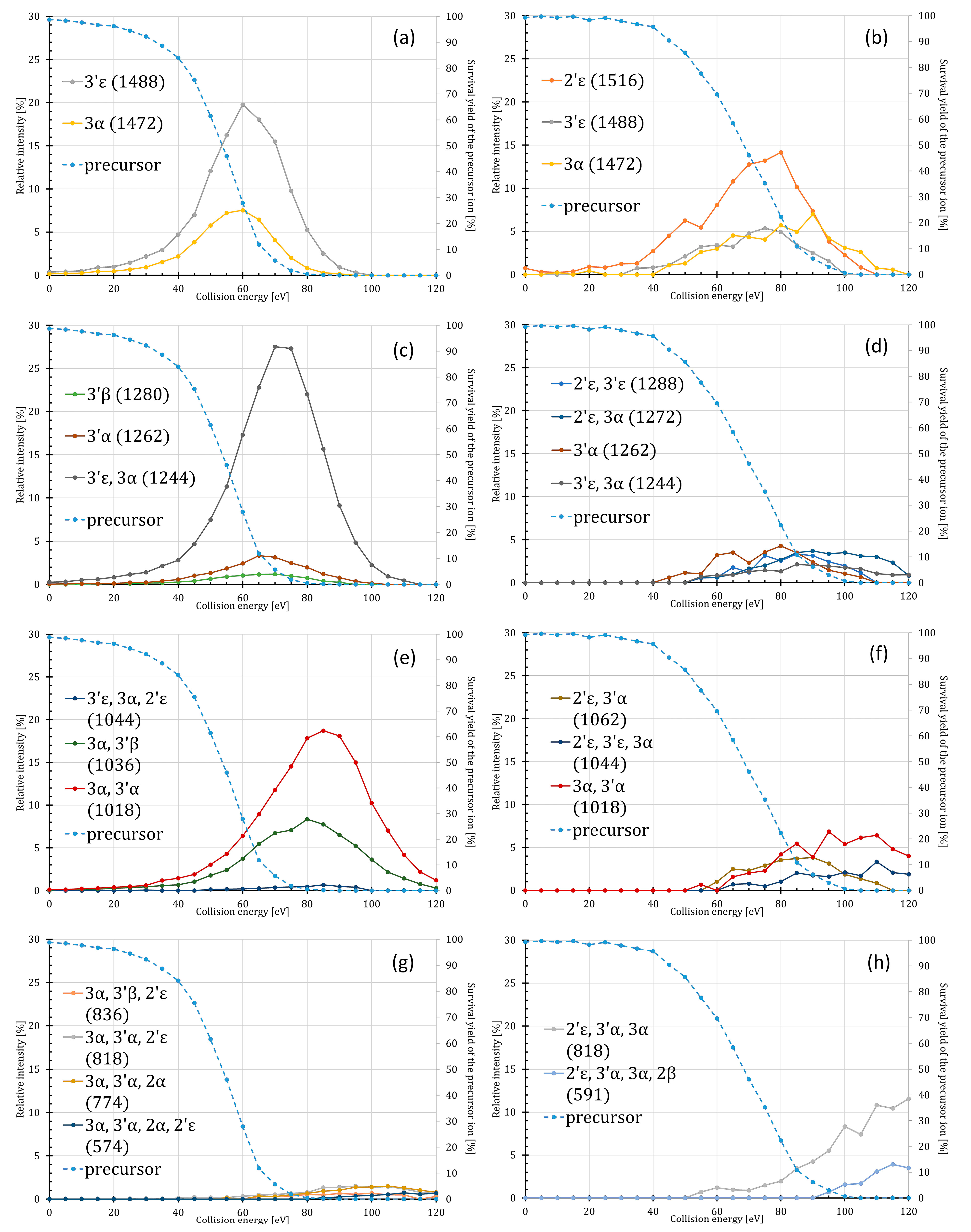
2.1. Energy Dependence of the Fatty Acyl Losses
2.2. Energy Dependence of the Intra- and Inter-Ring Fragmentations
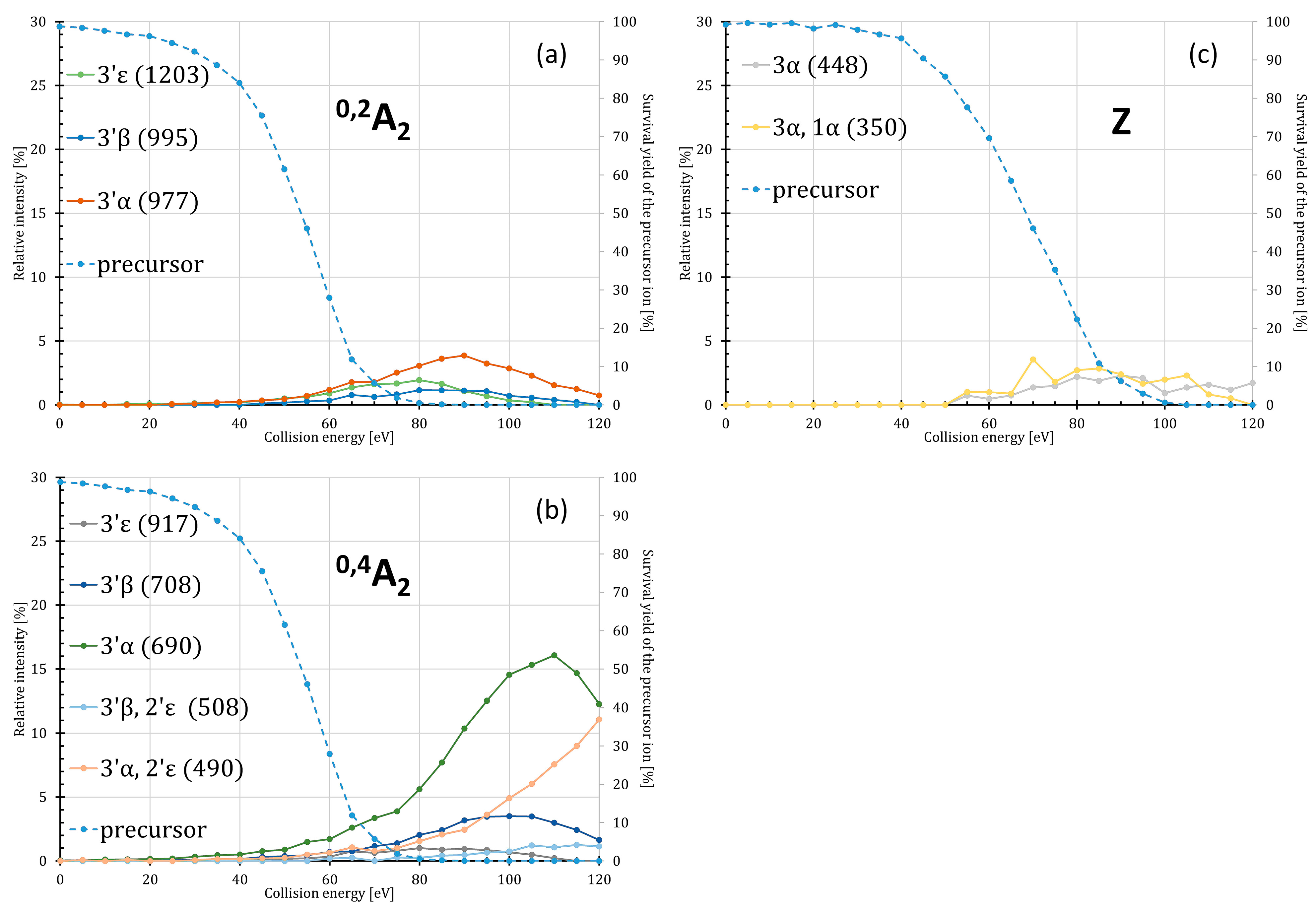
2.3. Energy Dependence of the Meta- and Orthophosphate Ion Losses
2.4. Identification of Chimera Mass Spectra of Lipid A Isomers
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Lipid A Isolation and Sample Preparation
4.3. High-Pressure Liquid Chromatography Coupled with Electrospray Ionization-Quadrupole-Time-of-Flight Tandem Mass Spectrometry
4.4. ERMS Data Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beutler, B.; Rietschel, E.T. Innate immune sensing and its roots: The story of endotoxin. Nat. Rev. Immunol. 2003, 3, 169–176. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef]
- Molinaro, A.; Holst, O.; Di Lorenzo, F.; Callaghan, M.; Nurisso, A.; D’Errico, G.; Zamyatina, A.; Peri, F.; Berisio, R.; Jerala, R.; et al. Chemistry of Lipid A: At the Heart of Innate Immunity. Chem.-Eur. J. 2015, 21, 500–519. [Google Scholar] [CrossRef]
- Trent, M.S.; Stead, C.M.; Tran, A.X.; Hankins, J.V. Diversity of endotoxin and its impact on pathogenesis. J. Endotoxin Res. 2006, 12, 205–223. [Google Scholar] [CrossRef]
- Wang, X.Y.; Quinn, P.J. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog. Lipid Res. 2010, 49, 97–107. [Google Scholar] [CrossRef]
- Miller, S.I.; Ernst, R.K.; Bader, M.W. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 2005, 3, 36–46. [Google Scholar] [CrossRef]
- Steimle, A.; Autenrieth, I.B.; Frick, J.S. Structure and function: Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. [Google Scholar] [CrossRef]
- Kussak, A.; Weintraub, A. Quadrupole ion-trap mass spectrometry to locate fatty acids on lipid A from Gram-negative bacteria. Anal. Biochem. 2002, 307, 131–137. [Google Scholar] [CrossRef]
- Corsaro, M.M.; Dal Piaz, F.; Lanzetta, R.; Parrilli, M. Lipid A structure of Pseudoalteromonas haloplanktis TAC 125: Use of electrospray ionization tandem mass spectrometry for the determination of fatty acid distribution. J. Mass Spectrom. 2002, 37, 481–488. [Google Scholar] [CrossRef]
- Madalinski, G.; Fournier, F.; Wind, F.L.; Afonso, C.; Tabet, J.C. Gram-negative bacterial lipid A analysis by negative electrospray ion trap mass spectrometry: Stepwise dissociations of deprotonated species under low energy CID conditions. Int. J. Mass Spectrom. 2006, 249, 77–92. [Google Scholar] [CrossRef]
- Silipo, A.; De Castro, C.; Lanzetta, R.; Molinaro, A.; Parrilli, M.; Vago, G.; Sturiale, L.; Messina, A.; Garozzo, D. Structural characterizations of lipids A by MS/MS of doubly charged ions on a hybrid linear ion trap/orbitrap mass spectrometer. J. Mass Spectrom. 2008, 43, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Crittenden, C.M.; Akin, L.D.; Morrison, L.J.; Trent, M.S.; Brodbelt, J.S. Characterization of Lipid A Variants by Energy-Resolved Mass Spectrometry: Impact of Acyl Chains. J. Am. Soc. Mass Spectrom. 2017, 28, 1118–1126. [Google Scholar] [CrossRef]
- Aissa, I.; Kilár, A.; Dörnyei, Á. Study on the CID Fragmentation Pathways of Deprotonated 4‘-Monophosphoryl Lipid A. Molecules 2021, 26, 5961. [Google Scholar] [CrossRef]
- Jones, J.W.; Cohen, I.E.; Turecek, F.; Goodlett, D.R.; Ernst, R.K. Comprehensive Structure Characterization of Lipid A Extracted from Yersinia pestis for Determination of its Phosphorylation Configuration. J. Am. Soc. Mass Spectrom. 2010, 21, 785–799. [Google Scholar] [CrossRef]
- Bedoux, G.; Vallee-Rehel, K.; Kooistra, O.; Zähringer, U.; Haras, D. Lipid A components from Pseudomonas aeruginosa PAO1 (serotype O5) and mutant strains investigated by electrospray ionization ion-trap mass spectrometry. J. Mass Spectrom. 2004, 39, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Kaltashov, I.A.; Doroshenko, V.; Cotter, R.J.; Takayama, K.; Qureshi, N. Confirmation of the structure of lipid A derived from the lipopolysaccharide of Rhodobacter sphaeroides by a combination of MALDI, LSIMS, and tandem mass spectrometry. Anal. Chem. 1997, 69, 2317–2322. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.J.; Altman, E. Structural characterization of the lipid A region of Aeromonas salmonicida subsp salmonicida lipopolysaccharide. Carbohydr. Res. 2006, 341, 2816–2825. [Google Scholar] [CrossRef]
- Phillips, N.J.; Adin, D.M.; Stabb, E.V.; McFall-Ngai, M.J.; Apicella, M.A.; Gibson, B.W. The Lipid A from Vibrio fischeri Lipopolysaccharide. A Unique Structure Bearing a Phosphoglycerol Moiety. J. Biol. Chem. 2011, 286, 21203–21219. [Google Scholar] [CrossRef] [PubMed]
- Casabuono, A.C.; van der Ploeg, C.A.; Roge, A.D.; Bruno, S.B.; Couto, A.S. Characterization of lipid A profiles from Shigella flexneri variant X lipopolysaccharide. Rapid Commun. Mass Spectrom. 2012, 26, 2011–2020. [Google Scholar] [CrossRef]
- Yang, H.; Smith, R.D.; Chandler, C.E.; Johnson, J.K.; Jackson, S.N.; Woods, A.S.; Scott, A.J.; Goodlett, D.R.; Ernst, R.K. Lipid A Structural Determination from a Single Colony. Anal. Chem. 2022, 94, 7460–7465. [Google Scholar] [CrossRef]
- Murphy, R.C.; Raetz, C.R.H.; Reynolds, C.M.; Barkley, R.M. Mass spectrometry advances in lipidomica: Collision-induced decomposition of Kdo(2)-lipid A. Prostaglandins Other Lipid Mediat. 2005, 77, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.A.; Cullen, T.W.; Trent, M.S.; Brodbelt, J.S. IR and UV Photodissociation as Analytical Tools for Characterizing Lipid A Structures. Anal. Chem. 2011, 83, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- El-Aneed, A.; Banoub, J. Elucidation of the molecular structure of lipid A isolated from both a rough mutant and a wild strain of Aeromonas salmonicida lipopolysaccharides using electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1683–1695. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Reinhold, V.N. Detailed Structural Characterisation of Lipid-A: Electrospray Ionization Coupled with Tandem Mass-Spectrometry. Anal. Biochem. 1994, 218, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.W.; Shaffer, S.A.; Ernst, R.K.; Goodlett, D.R.; Turecek, F. Determination of pyrophosphorylated forms of lipid A in Gram-negative bacteria using a multivaried mass spectrometric approach. Proc. Natl. Acad. Sci. USA 2008, 105, 12742–12747. [Google Scholar] [CrossRef] [PubMed]
- Sándor, V.; Dörnyei, Á.; Makszin, L.; Kilár, F.; Peterfi, Z.; Kocsis, B.; Kilár, A. Characterization of complex, heterogeneous lipid A samples using HPLC-MS/MS technique I. Overall analysis with respect to acylation, phosphorylation and isobaric distribution. J. Mass Spectrom. 2016, 51, 1043–1063. [Google Scholar] [CrossRef] [PubMed]
- Sándor, V.; Kilár, A.; Kilár, F.; Kocsis, B.; Dörnyei, Á. Characterization of complex, heterogeneous lipid A samples using HPLC-MS/MS technique II. Structural elucidation of non-phosphorylated lipid A by negative-ion mode tandem mass spectrometry. J. Mass Spectrom. 2016, 51, 615–628. [Google Scholar] [CrossRef]
- Sándor, V.; Kilár, A.; Kilár, F.; Kocsis, B.; Dörnyei, Á. Characterization of complex, heterogeneous lipid A samples using HPLC-MS/MS technique III. Positive-ion mode tandem mass spectrometry to reveal phosphorylation and acylation patterns of lipid A. J. Mass Spectrom. 2018, 53, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Sándor, V.; Berkics, B.V.; Kilár, A.; Kocsis, B.; Kilár, F.; Dörnyei, Á. NACE-ESI-MS/MS method for separation and characterization of phosphorylation and acylation isomers of lipid A. Electrophoresis 2020, 41, 1178–1188. [Google Scholar] [CrossRef]
- Sándor, V.; Űrmös, B.; Aissa, I.; Dörnyei, Á.; Kilár, A. Characterization of isomeric lipid A species from Pseudomonas aeruginosa PAO1 by non-aqueous capillary electrophoresis with positive and negative ion electrospray tandem mass spectrometry. Arab. J. Chem. 2023, 16, 104944–104953. [Google Scholar] [CrossRef]
- Almostafa, M.; Allehyane, B.; Egli, S.; Bottaro, C.; Fridgen, T.D.; Banoub, J. Tandem mass spectrometry determination of the putative structure of a heterogeneous mixture of Lipid As isolated from the lipopolysaccharide of the Gram-negative bacteria Aeromonas liquefaciens SJ-19a. Rapid Commun. Mass Spectrom. 2016, 30, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Lukasiewicz, J.; Jachymek, W.; Niedziela, T.; Kenne, L.; Lugowski, C. Structural analysis of the lipid A isolated from Hafnia alvei 32 and PCM 1192 lipopolysaccharides. J. Lipid Res. 2010, 51, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Tawab, A.; Akbar, N.; Hasssan, M.; Habib, F.; Ali, A.; Rahman, M.; Jabbar, A.; Rauf, W.; Iqbal, M. Mass spectrometric analysis of lipid A obtained from the lipopolysaccharide of Pasteurella multocida. RSC Adv. 2020, 10, 30917–30933. [Google Scholar] [CrossRef] [PubMed]
- Froning, M.; Helmer, P.O.; Hayen, H. Identification and structural characterization of lipid A from Escherichia coli, Pseudomonas putida and Pseudomonas taiwanensis using liquid chromatography coupled to high-resolution tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2020, 34, e8897. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.J.; Parker, W.R.; Holden, D.D.; Henderson, J.C.; Boll, J.M.; Trent, M.S.; Brodbelt, J.S. UVliPiD: A UVPD-Based Hierarchical Approach for De Novo Characterization of Lipid A Structures. Anal. Chem. 2016, 88, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Costello, C.E. A Systematic Nomenclature for Carbohydrate Fragmentations in FAB-MS MS Spectra of Glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Okahashi, N.; Ueda, M.; Matsuda, F.; Arita, M. Analyses of Lipid A Diversity in Gram-Negative Intestinal Bacteria Using Liquid Chromatography-Quadrupole Time-of-Flight Mass Spectrometry. Metabolites 2021, 11, 197. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; McLendon, M.K.; Phillips, N.J.; Apicella, M.A.; Gibson, B.W. Characterization of lipid A acylation patterns in Francisella tularensis, Francisella novicida, and Francisella philomiragia using multiple-stage mass spectrometry and matrix-assisted laser desorption/ionization on an intermediate vacuum source linear ion trap. Anal. Chem. 2007, 79, 1034–1042. [Google Scholar] [CrossRef]
- Westphal, O.; Lüderitz, O.; Bister, F. Über die Extraktion von Bakterien mit Phenol Wasser. Z. Naturforschung Sect. B 1952, 7, 148–155. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cleavage Type # | Elemental Composition of the Loss | Cleavage Site(s) | Monoisotopic Molecular Mass of the Loss | Monoisotopic Ion Mass of the Product Ion |
|---|---|---|---|---|
| One acyl chain | ||||
| C12:0 | C12H24O2 | 2′ε | 200.1776 | 1516.0682 * |
| C14:0 | C14H28O2 | 3′ε | 228.2089 | 1488.0369 |
| C14:0(3-OH) | C14H28O3 | 3α | 244.2038 | 1472.0419 |
| Two acyl chains | ||||
| C12:0 and C14:0 | C26H52O4 | 2′ε, 3′ε | 428.3866 | 1287.8592 * |
| C14:0(3-O-C14:0)ketene | C28H52O3 | 3′β | 436.3916 | 1279.8541 ** |
| C12:0 and C14:0(3-OH) | C26H52O5 | 2′ε, 3α | 444.3815 | 1271.8643 |
| C14:0(3-O-C14:0) | C28H54O4 | 3′α | 454.4022 | 1261.8436 |
| C14:0 and C14:0(3-OH) | C28H56O5 | 3′ε, 3α | 472.4128 | 1243.8330 |
| Three acyl chains | ||||
| C12:0 and C14:0(3-O-C14:0) | C40H78O6 | 2′ε, 3′α | 654.5798 | 1061.6659 * |
| C12:0, C14:0, and C14:0(3-OH) | C40H80O7 | 2′ε, 3′ε, 3α | 672.5904 | 1043.6554 |
| C14:0(3-OH) and C14:0(3-O-C14:0)ketene | C42H80O6 | 3α, 3′β | 680.5955 | 1035.6503 ** |
| C14:0(3-OH) and C14:0(3-O-C14:0) | C42H82O7 | 3α, 3′α | 698.6061 | 1017.6397 |
| Four acyl chains | ||||
| C12:0, C14:0(3-OH), and C14:0(3-O-C14:0)ketene | C54H104O8 | 2′ε, 3′β, 3α | 880.7731 | 835.4727 ** |
| C12:0, C14:0(3-OH), and C14:0(3-O-C14:0) | C54H106O9 | 2′ε, 3′α, 3α | 898.7837 | 817.4621 |
| C14:0(3-OH), C14:0(3-O-C14:0), and C14:0(3-OH)amide | C56H111N1O9 | 3α, 3′α, 2α | 941.8259 | 774.4199 ** |
| Five acyl chains | ||||
| C12:0, C14:0(3-OH), C14:0(3-O-C14:0), and C12:0aldehyde | C66H130O10 | 2′ε, 3′α, 3α, 2δ | 1082.9664 | 633.2794 * |
| C12:0, C14:0(3-OH), C14:0(3-O-C14:0), and C14:0(3-OH)ketene | C68H132O11 | 2′ε, 3′α, 3α, 2β | 1124.9770 | 591.2688 * |
| C14:0(3-OH), C14:0(3-O-C14:0), C14:0(3-OH)amide, and C12:0 | C68H135N1O11 | 3α, 3′α, 2α, 2′ε | 1142.0035 | 574.2423 ** |
| Cleavage Type # | Elemental Composition of the Loss | Cleavage Site(s) | Monoisotopic Molecular Mass of the Loss | Monoisotopic Ion Mass of the Product Ion |
|---|---|---|---|---|
| 0,2A2-type ions for P4′ | C16H31NO3 | 0,2A2 | 285.2304 | 1431.0154 * |
| C14:0 | C30H59NO5 | 0,2A2, 3′ε | 513.4393 | 1202.8065 |
| C14:0(3-O-C14:0)ketene | C44H83NO6 | 0,2A2, 3′β | 721.6220 | 994.6237 |
| C14:0(3-O-C14:0) | C44H85NO7 | 0,2A2, 3′α | 739.6326 | 976.6132 |
| 0,4A2-type ions for P4′ | C32H61NO7 | 0,4A2 | 571.4448 | 1144.8010 * |
| C14:0 | C46H89NO9 | 0,4A2, 3′ε | 799.6537 | 916.5921 |
| C14:0(3-O-C14:0)ketene | C60H113NO10 | 0,4A2, 3′β | 1007.8364 | 708.4093 |
| C14:0(3-O-C14:0) | C60H115NO11 | 0,4A2, 3′α | 1025.8470 | 690.3988 |
| C12:0 and C14:0(3-O-C14:0)ketene | C72H137NO12 | 0,4A2, 3′β, 2′ε | 1208.0141 | 508.2317 |
| C12:0 and C14:0(3-O-C14:0) | C72H139NO13 | 0,4A2, 3′α, 2′ε | 1226.0246 | 490.2211 |
| B-type ions for P4′ | C34H65NO9 | B1 | 631.4659 | 1084.7799 * |
| C14:0(3-O-C14:0)ketene | C62H117NO12 | B1, 3′β | 1067.8576 | 648.3882 |
| C14:0(3-O-C14:0) | C62H119NO13 | B1, 3′α | 1085.8681 | 630.3776 |
| C12:0 and C14:0(3-O-C14:0)ketene | C74H141NO14 | B1, 3′β, 2′ε | 1268.0352 | 448.2106 |
| C12:0 and C14:0(3-O-C14:0) | C74H143NO15 | B1, 3′α, 2′ε | 1286.0458 | 430.2000 |
| Z-type ions for P1 | C60H113NO11 | Z1 | 1023.83136 | 692.4144 # |
| C14:0(3-OH) | C74H141NO14 | Z1, 3α | 1268.0352 | 448.2106 |
| C14:0(3-OH) and H3PO4 | C74H144NO18P | Z1, 3α, 1α | 1366.0121 | 350.2337 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dörnyei, Á.; Kilár, A.; Sándor, V. Identification of a Chimera Mass Spectrum of Isomeric Lipid A Species Using Negative Ion Tandem Mass Spectrometry. Toxins 2024, 16, 322. https://doi.org/10.3390/toxins16070322
Dörnyei Á, Kilár A, Sándor V. Identification of a Chimera Mass Spectrum of Isomeric Lipid A Species Using Negative Ion Tandem Mass Spectrometry. Toxins. 2024; 16(7):322. https://doi.org/10.3390/toxins16070322
Chicago/Turabian StyleDörnyei, Ágnes, Anikó Kilár, and Viktor Sándor. 2024. "Identification of a Chimera Mass Spectrum of Isomeric Lipid A Species Using Negative Ion Tandem Mass Spectrometry" Toxins 16, no. 7: 322. https://doi.org/10.3390/toxins16070322